Bases moleculares de la esclerosis tuberosa
La esclerosis tuberosa es un síndrome congénito neurocutáneo (o facomatosis) que se caracteriza por el desarrollo generalizado de hamartomas en múltiples órganos. Para las personas afectadas, las complicaciones neurológicas y psiquiátricas son las características más discapacitantes y mortales. Aunque su fenotipo clínico es complejo, sólo tres lesiones caracterizan las características neuropatológicas de la enfermedad: tubérculos corticales, nódulos subependimarios y astrocitomas subependimarios de células gigantes. Este último, es un tumor benigno en el cerebro de origen mixto neuronal y glial.
Células gigantes y esclerosis tuberosa
A las lesiones cerebrales corticales focales (tubérculos), reconocidas por primera vez por Bourneville en 1879 durante el examen post-mortem de una niña de 15 años de edad, se les dio el nombre de esclerosis tuberosa de las circunvoluciones cerebrales. Posteriores estudios clínicos definieron al complejo de esclerosis tuberosa (CET o TSC, por su sigla en inglés) como un trastorno autosómico dominante hereditario en la que los tumores se desarrollan en múltiples órganos y sistemas. Los análisis patológicos pudieron identificar células gigantes en los tubérculos corticales y en los tumores cerebrales, conocidos como astrocitomas subependimarios de células gigantes, que se encuentran con frecuencia en los pacientes con CET.
La base molecular de la formación de células gigantes en la esclerosis tuberosa permaneció en la oscuridad hasta la década de 1990, incluso después del descubrimiento de los dos genes TSC1 y TSC2, los causantes de este trastorno. En 2001, el cribado genético de mutaciones que aumentan en el tamaño de las células oculares en Drosophila condujo al descubrimiento que la pérdida de TSC1 o TSC2 produce un fenotipo de células grandes. Estudios genéticos y funcionales posteriores han demostrado que TSC1 y TSC2, junto con una tercera proteína, la TBC1D7, forman el complejo de proteína de CET, que funciona como regulador negativo de la diana de rapamicina en células de mamífero (mTOR) complejo 1 (mTORC1) (fig. 1). Se ha descrito en detalle la vía de mTOR, dado su papel fundamental en la regulación del tamaño celular y el crecimiento, junto a su importancia en diversos tipos de cáncer y numerosos procesos fisiológicos. La pérdida de TSC1 o TSC2 se produce en los tumores CET para complementar una mutación germinal en un gen o en otro, lo que conduce a la activación constitutiva de mTORC1 y a procesos anabólicos, incluyendo la síntesis de proteínas, lípidos y ácidos nucleicos.
Se requieren aumentos en el número y tamaño celular para el crecimiento normal (durante el desarrollo o reparación de la lesión) y el crecimiento anormal (por ejemplo, cáncer). Las células deben duplicarse a través de la biosíntesis macromolecular de todos sus componentes para permitir la división celular sin una pérdida neta de tamaño. Sobre la base de los estudios que muestran que muchos genes pueden influir en el tamaño celular en levaduras y Drosophila, Diane C. Fingar y colaboradores (Genes Dev 2002; 16:1472-87) exploraron el papel de mTOR en la regulación del tamaño celular, observando que el inhibidor de mTORC1 (rapamicina, ahora conocida como sirolimus) reduce la dimensión celular. Este efecto es bloqueado por un mutante de mTOR resistente a la rapamicina, y parcialmente y de forma sinérgica mediante la expresión de un mutante de la quinasa S6 1 (S6K1) resistente a la rapamicina y por la expresión del factor de iniciación de la traducción eucariótica 4E (EIF4E) (fig. 1). Estos resultados son consistentes con la identificación de S6K1 y la proteína de unión 1 del factor de iniciación de la traducción eucariota 4E (4EBP1) como los principales objetivos de la regulación por mTORC1, con 4EBP1 actuando como un inhibidor de la traducción a través de la unión a EIF4E, que juntos permiten un aumento del tamaño celular en preparación para la división celular.
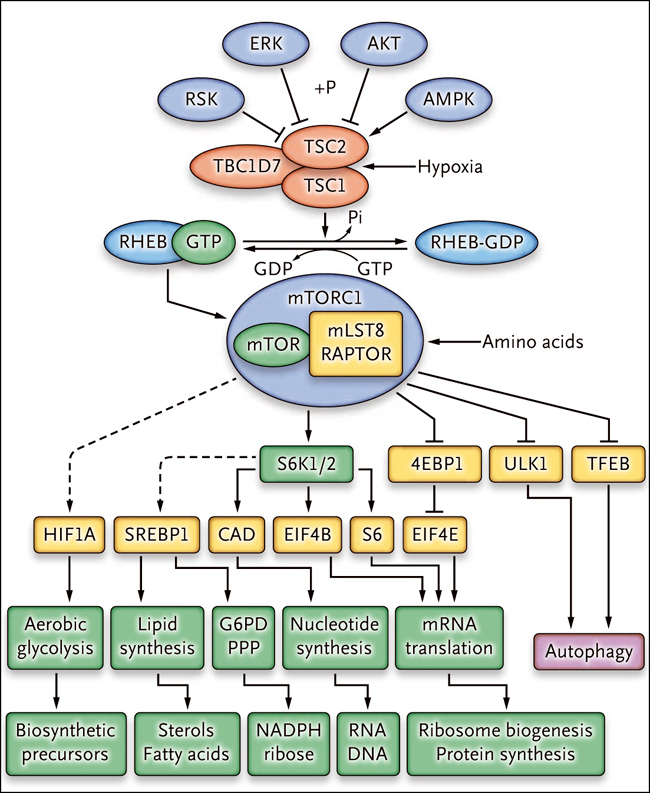
Figura 1: complejo de proteínas TSC, complejo1 mTOR y sus efectos
Las proteínas del complejo de esclerosis tuberosa (TSC, por sus siglas en inglés), que incluyen TSC1, TSC2 y TBC1D7, están reguladas negativamente por fosforilación (+P) vía AKT, ERK y RSK, que son quinasas fundamentales que se activan durante la señalización de crecimiento. Este complejo se activa en condiciones de hipoxia y durante la fosforilación por AMPK en respuesta al estrés energético y determinadas funciones. Las flechas continuas indican acontecimientos estimulantes, que a veces están mediados por la fosforilación; las líneas intercaladas denotan efectos inhibitorios.
¿Cómo afecta la activación de mTORC1 al aumento celular? Lo hace a través de varios mecanismos que controlan el metabolismo anabólico, entre los cuales está el incremento en la síntesis de proteínas por la generación de ribosomas adicionales y mayores tasas de traducción del ARN mensajero (fig. 1). El complejo mTORC1 regula la traducción de un conjunto de ARN mensajeros que tienen una secuencia de oligopirimidina en la región terminal 5' (5'-TOP) por fosforilación directa de las proteínas 4EBP, provocando su liberación a partir de EIF4E. Estos ARN mensajeros 5'-TOP se enriquecen de los que codifican proteínas ribosomales y factores de traducción, aumentando la biogénesis y la síntesis de proteínas del ribosoma. La activación de mTORC1 incrementa la traducción del factor inducible por hipoxia 1α (HIF1A) para promover la expresión de los transportadores de glucosa y las enzimas glucolíticas, y así estimular la glicolisis aerobia, que proporciona mediadores glicolíticos para la síntesis de lípidos, aminoácidos y nucleótidos. También activa las proteínas de unión al elemento regulador de esteroles (SREBP1 y SREBP2) para mejorar la expresión de las enzimas necesarias para la biosíntesis de ácidos grasos y esteroles. Además, mTORC1 promueve la expresión de genes en la ruta de la pentosa fosfato para optimizar la producción de NADPH, que es necesaria para la biosíntesis lipídica. También incita la biosíntesis de pirimidina a través de la fosforilación mediada por S6K1 de la enzima CAD (carbamil fosfato sintetasa II, aspartato transcarbamilasa, dihidroorotasa) para aumentar la producción de nucleótidos de ARN y ADN. Al mismo tiempo, mTORC1 inhibe fuertemente la autofagia para preservar orgánulos celulares y macromoléculas por fosforilación del factor de transcripción TFEB y de ULK1 (conocido como ATG1), una proteína quinasa que regula la iniciación autofagosomas.
La evidencia de la activación de la vía de mTOR se ha visto en las células gigantes de tubérculos y en astrocitomas subependimarios de células gigantes. Esta comprensión del mecanismo básico de formación de células gigantes y el crecimiento tumoral en el CET ha llevado al uso de sirolimus y otros inhibidores de mTOR para el tratamiento de sus diversas manifestaciones.
Varios tumores CET o enfermedades relacionadas, incluyendo el angiomiolipoma renal, la linfangioleiomiomatosis y el astrocitoma subependimario de células gigantes, son sensibles al tratamiento con inhibidores de mTOR (sirolimus y everolimus). Además, en algunos pacientes con cáncer de riñón y de vejiga con mutaciones TSC1 que no tienen CET, los tumores han sido sensibles a la inhibición de mTORC1. El everolimus genera una reducción volumétrica en los tumores CET de aproximadamente un 50% y, ocasionalmente, destruye los tumores, lo que sugiere que algunas de las respuestas pueden incluir más de una disminución en el tamaño celular. Los estudios en ratones han demostrado que las células que carecen de CET tienen una importante reducción de tamaño en respuesta a la terapia de inhibición de mTOR in vivo, un resultado que probablemente sea por el cese de la estimulación anabólica y la inducción de autofagia. La mayoría de los angiomiolipomas renales vuelven a crecer, en gran parte cuando se termina el tratamiento, y los astrocitomas subependimarios de células gigantes también pueden volver a crecer cuando se interrumpe la terapia, indicando que la continuación del tratamiento es necesaria para mantener una respuesta. Afortunadamente, los efectos metabólicos y anabólicos de la activación de mTORC1 que impulsan un aumento en el tamaño celular ofrecen abundantes oportunidades para un ataque terapéutico adicional, controlando eficazmente la enfermedad a largo plazo e incluso su eliminación.
La descripción clínica de CET y los estudios patológicos que han identificado las células gigantes estimulan la investigación de la regulación del tamaño celular y el crecimiento. Esta investigación ha generado avances clínicos que benefician a los pacientes con esclerosis tuberosa.
Fuente bibliográfica
Molecular Basis of Giant Cells in Tuberous Sclerosis Complex
David J. Kwiatkowski, M.D., Ph.D., and Brendan D. Manning, Ph.D.
Department of Medicine, Brigham and Women’s Hospital (D.J.K.), and the Department of Genetics and Complex Diseases, Harvard School of Public Health (B.D.M.) — both in Boston.
DOI: 10.1056/NEJMcibr1406613