Autofagia, p53 y el cáncer de páncreas
La macroautofagia (autofagia) es una ruta catabólica regulada para degradar organelos celulares y macromoléculas. Su papel en el cáncer es complejo y puede variar según el tipo de tumor o contexto. Se sabe que los tumores primarios del cáncer de páncreas muestran una elevada autofagia bajo condiciones basales. Su inhibición genética o farmacológica lleva a un aumento de las especies reactivas de oxígeno, daño del ADN y un defecto metabólico que conduce a una disminución de la fosforilación oxidativa mitocondrial. Todo, en última instancia, resulta en la supresión del crecimiento celular del cáncer de pancreático. Lo más importante, la inhibición de la autofagia por medios genéticos o tratamiento con cloroquina conduce a la regresión del tumor y prolonga la supervivencia en xenoinjertos de cáncer de páncreas y en modelos genéticos de ratón.
Estos resultados proponen que, a diferencia de otros tipos de neoplasias en el que la inhibición de la autofagia sinergiza la quimioterapia para prevenir su sobre-regulación como mecanismo de supervivencia reactiva, la autofagia es realmente necesaria para el crecimiento tumorigénico de los cánceres pancreáticos, y aquellos medicamentos que la inactiven podrían tener una utilidad clínica en el tratamiento de los cánceres de páncreas y de otros tumores malignos con dependencia similar.
Desarrollo de tumores pancreáticos y autofagia
En esta era de la conciencia ambiental y el ser verde, se puede aprender una lección de las células eucariotas. Un proceso eficiente llamado autofagia implica la formación de vesículas intracelulares, llamadas autofagosomas, que entrega proteínas y orgánulos de larga vida a los lisosomas para su degradación. Las macromoléculas de esta degradación se utilizan en una variedad de procesos celulares, entonces, no es sorprendente que la desregulación de la autofagia juegue un papel importante en varias enfermedades humanas, incluyendo el cáncer. Sin embargo, si la autofagia promueve o suprime una neoplasia es tema de debate. La evidencia reciente reportada por Mathias T. Rosenfeldt y colaboradores (Nature 2013; 504:296-300) aclara el rol del proceso en un modelo de ratón de adenocarcinoma ductal pancreático y su íntima vinculación con el estado mutacional de Trp53, gen del animal que codifica el homólogo de la proteína p53 supresora de tumores en humanos.
En el modelo utilizado por los investigadores, el oncogén Kras se activa en el compartimento epitelial del páncreas. Ellos encontraron que la inactivación condicional de ATG7, un mediador clave de la autofagia, obstruye la progresión del adenocarcinoma ductal pancreático. Este bloqueo se debe al aumento de muerte celular, detención del crecimiento y a la senescencia de las primeras etapas de las lesiones neoplásicas intraepiteliales pancreáticas. Por el contrario, la inactivación de ATG7 en el contexto de coexistencia entre Kras mutante y la supresión Trp53, no sólo no bloquea la progresión a adenocarcinoma invasivo ductal pancreático sino que también la mejora. La inhibición farmacológica de la autofagia por hidroxicloroquina en estos ratones produce los mismos efectos que la supresión de ATG7, lo que indica que la supresión del tumor frente a su activación puede estar relacionada con el estado funcional de Trp53.
Los presentes hallazgos tienen tres implicaciones clínicas inmediatas. En primer lugar, dado que la activación de KRAS es un evento temprano en la carcinogénesis pancreática humana, en donde la inactivación de TP53 es un acontecimiento tardío, la inhibición de la autofagia puede prevenir el desarrollo del adenocarcinoma ductal pancreático (Figura 1). Este enfoque es particularmente relevante para los pacientes con mayor riesgo de padecer la enfermedad.
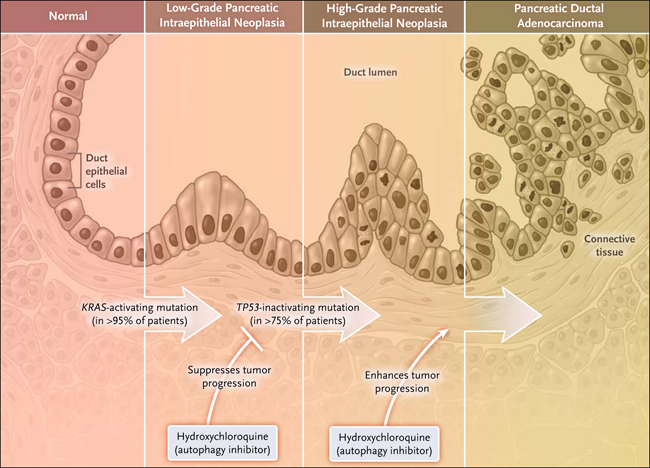
Figura 1: genética de la carcinogénesis de páncreas
El desarrollo de mutaciones KRAS en células epiteliales normales, que se encuentran en más del 95% de los pacientes con carcinoma pancreático, conduce a la formación de lesiones neoplásicas intraepiteliales pancreáticas de bajo grado. Con el tiempo, la acumulación de mutaciones adicionales, tales como en el gen que codifica la proteína supresora de tumores p53 (TP53), que se encuentran en más de 75% de los pacientes con tumores, conduce al desarrollo de lesiones pancreáticas neoplásicas intraepiteliales y cáncer de páncreas de alto grado. En este estudio, los autores utilizaron un modelo transgénico de adenocarcinoma ductal pancreático para demostrar que la inhibición de la autofagia bloquea la progresión de las lesiones neoplásicas intraepiteliales pancreáticas de bajo grado y del cáncer en ratones con Trp53 normal, mientras que la inhibición de la autofagia en presencia de mutaciones Trp53 promueve la formación neoplásica. Estas observaciones señalan que la caracterización de TP53 ayudará a identificar a los pacientes con diagnóstico reciente de adenocarcinoma ductal de páncreas que pueden beneficiarse de los inhibidores de la autofagia.
En segundo lugar, el tratamiento de pacientes con cáncer de páncreas que reciben inhibidores farmacológicos de la autofagia, como la hidroxicloroquina, en el contexto de la coexistencia de KRAS y de mutaciones TP53, en realidad puede estimular la progresión tumoral. Aproximadamente el 75% de los adenocarcinomas ductales pancreáticos tiene la coexistencia KRAS y alteraciones de TP53, y actualmente hay cinco ensayos clínicos activos que evalúan el papel de la hidroxicloroquina en el cáncer de páncreas resecable y avanzado. Por el contrario, los individuos seleccionados, con la condición TP53 intacta se pueden beneficiar de los inhibidores de la autofagia como un tratamiento adyuvante o de primera línea. La caracterización genética de los adenocarcinomas ductales pancreáticos antes del tratamiento puede permitir una mejor selección de regímenes terapéuticos específicos y agresivos, en donde los métodos de tratamiento combinado con el uso de estos inhibidores también proporcionan una oportunidad para la cura.
En tercer lugar, estos hallazgos podrían explicar datos controvertidos que sugieren que el aumento de avidez de la PET, medida funcional del metabolismo evaluado por medio de la tomografía por emisión de positrones (PET), se correlaciona con una menor supervivencia en los pacientes con cáncer pancreático localmente avanzado. Por ejemplo, el equipo de Mathias T. Rosenfeldt encontró que en adenocarcinomas con Trp53 intacto, la pérdida de la autofagia genera una disminución en el consumo de oxígeno y en el metabolismo, mientras que en adenocarcinomas sin Trp53, la pérdida de la autofagia causa un aumento en el consumo de glucosa. Este descubrimiento sugiere que puede existir una base genética o molecular en las variaciones de avidez de la PET. Una mejor comprensión de la interacción genética, la autofagia, el metabolismo del tumor y la avidez de PET o de otra imagen funcional, permitiría la selección de pacientes con carcinomas con TP53 no mutado, con más probabilidades de beneficiarse de los inhibidores de la autofagia que aquellos con tumores con formas mutadas de TP53.
Fuente bibliográfica
Autophagy, p53, and Pancreatic Cancer
Christine A. Iacobuzio-Donahue, M.D., Ph.D., and Joseph M. Herman, M.D.
Department of Pathology and the David Rubenstein Center for Pancreatic Cancer Research, Memorial Sloan-Kettering Cancer Center, New York (C.A.I.-D.); and the Department of Radiation Oncology and Molecular Radiation Sciences, Johns Hopkins Hospital, Baltimore (J.M.H.).
N Engl J Med 2014; 370:1352-1353