Transición maligna en la hematopoiesis
Las leucemias agudas de varios tipos se encuentran entre los trastornos sanguíneos más graves. La leucemia causa una severa enfermedad, con elevadas tasas de mortalidad. Una mejor comprensión de las causas de las distintas formas de esta patología podría llevar a un diagnóstico más temprano y un tratamiento más efectivo. Dentro de lo que se conoce, el gen ASXL1 a menudo se encuentra mutado en cánceres mieloides y también en la hematopoyesis clonal de potencial indeterminado (CHIP, por sus siglas en inglés) un trastorno que se caracteriza por mutaciones somáticas en células hematopoyéticas pero sin otros criterios para ser catalogado como cáncer hematológico. A su vez la CHIP se asocia con una mayor probabilidad de progresión a cánceres mieloides y linfoides. Por lo que su estudio enfocado en un diagnóstico temprano podría conferir a futuro mejores resultados a los pacientes.
Mutaciones en Asxl1
Diversas mutaciones en ASXL1 están localizadas en el último exón (codificando la región C-terminal de la proteína ASXL1) y son "dominantes-negativas " - es decir, resultan en una proteína mutante que inhibe la proteína ASXL1 normal codificada por el alelo silvestre. Nagase y sus colegas describieron recientemente un modelo de ratón de CHIP que diseñaron mediante el "knockout" condicional de una mutación Asxl1 en el último exón de Asxl1 (DOI: 10.1084/jem.20171151). La versión mutada de Asxl1 se expresó en la mayoría de las células hematopoyéticas en el ratón mutante, junto con Asxl1 de tipo silvestre; el ratón tenía características típicamente vistas en la CHIP, con inclinación mieloide, anemia dependiente de la edad, trombocitosis y displasia morfológica.
Los autores luego se preguntaron si la hematopoyesis clonal conduce a la leucemia. Los ratones tuvieron alteraciones en el epigenoma (las modificaciones bioquímicas del genoma o de las proteínas que componen la cromatina) con reducciones sustanciales en H3K4me3 (trimetilación de la lisina 4 en la proteína histona 3) y H2AK119Ub (ubiquitinación de la histona 2, que está relacionada con el silenciamiento del gen mediado por polycomb). La leucemia no se desarrolló en los ratones mutantes durante un período de observación de 18 meses.
Sin embargo, la sobreexpresión de Runx mutado en la médula ósea de los ratones mutantes Asxl1 fue seguida por el desarrollo de leucemia mieloide aguda y hepatoesplenomegalia después de un corto período de latencia. (La sobreexpresión de RUNX mutado ha sido identificada en pacientes con el síndrome mielodisplásico y leucemia). De manera similar, la mutagénesis insercional mediada por retrovirus se utilizó para generar mutaciones adicionales en los ratones mutantes Asxl1. Se desarrolló leucemia aguda en todos los ratones infectados (figura 1).
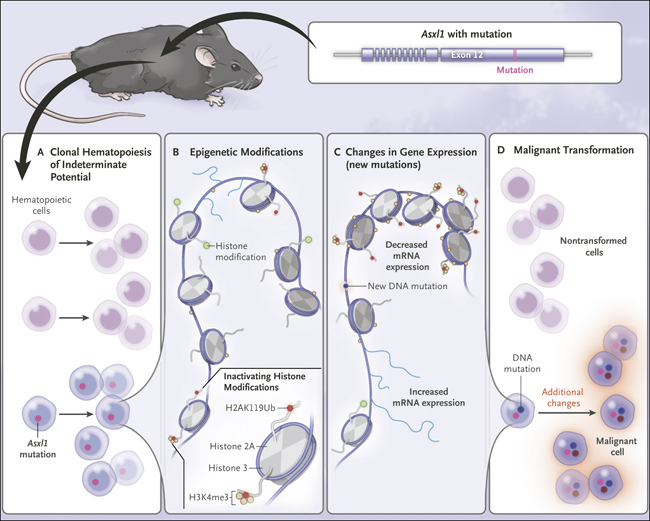
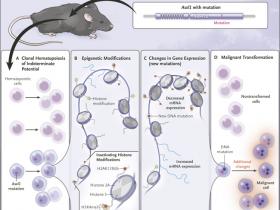
Figura 1. Modelando la transformación de la hematopoyesis clónica de potencial intermedio (CHIP) a la leucemia mieloide aguda.
Nagase y sus colegas describieron recientemente un modelo de ratón de CHIP y su transformación a un modelo de leucemia mieloide aguda. Una mutación en Asxl1 causa CHIP (panel A) y conduce a modificaciones epigenéticas con reducciones sustanciales en H3K4me3 y H2AK119Ub (panel B). Esto causa alteraciones en la expresión génica y puede conducir a nuevas mutaciones (panel C). Finalmente, las mutaciones y alteraciones adicionales en la expresión génica pueden conducir a una transformación maligna (panel D). El término ARNm denota ARN mensajero.
La hematopoyesis en el modelo de ratón de CHIP mostró varias anormalidades: un aumento en las células mieloides y células mieloides inmaduras y un modesto bloqueo en el desarrollo eritroide. En la médula ósea, se observó una reducción de las células madre hematopoyéticas a largo plazo y de las células progenitoras multipotentes. En un ensayo de trasplante competitivo, en el que se comparan células madre mutantes con células madre normales, las primeras mostraron una reducción del injerto en sangre periférica y médula ósea. Sin embargo, las células madre hematopoyéticas a largo plazo y las progenitoras multipotentes no se vieron afectadas, lo que lleva a la conclusión de que la supervivencia de las células madre hematopoyéticas no se ve afectada. Estos hallazgos pueden explicar en parte por qué se mantuvo el crecimiento clonal.
El diagnóstico de la hematopoyesis clonal implicaría idealmente la identificación del clon en expansión de células madre y progenitoras antes de la transformación al síndrome mielodisplásico y a la leucemia mielodisplásica aguda, de modo que se pudieran probar intervenciones para prevenir la transformación. Desafortunadamente, esto no es posible, debido a la falta de marcadores específicos en la superficie celular para identificar el clon en expansión. En consecuencia, los médicos actualmente se limitan a los factores hematológicos (alteración mieloide, anemia y trombocitosis) junto con la secuenciación de los genes candidatos (por ejemplo, TET2, DNMT3A y ASXL1) en las células hematopoyéticas.
De manera similar, una vez que el cáncer se ha desarrollado, un diagnóstico temprano se asocia con mejores resultados. No obstante, actualmente no es posible tratar el síndrome mielodisplásico y la leucemia mielodisplásica aguda a nivel de "precisión" porque se carece de fármacos que bloqueen específicamente la función de la proteína mutante (por ejemplo, ASXL1) en humanos.
Sin embargo, el cribaje (screening) es técnicamente posible, y si se diagnostica CHIP, los síntomas pueden ser tratados al menos parcialmente, y el paciente puede ser monitoreado para el desarrollo de cáncer. Pero debido a que la CHIP se encuentra en por lo menos el 10% de las personas de 70 a 80 años de edad, tal revisión sería un desafío. Aunque estas personas tienen un riesgo más alto de neoplasia hematológica (por un factor de aproximadamente 13) en comparación con la población en general, la frecuencia de transformación es de sólo 0,5 a 1% por año, lo que hace difícil justificar la evaluación con las herramientas actualmente disponibles.
Fuente bibliográfica
Tipping Clonal Hematopoiesis into Transformation
Stefan Karlsson, M.D., Ph.D.
Department of Molecular Medicine and Gene Therapy, Lund University, Lund, Sweden.
DOI: 10.1056/NEJMcibr1803641