Redescubriendo las hemoglobinopatías
Las hemoglobinopatías, trastornos causados por mutaciones en los genes que codifican globinas "adultas", ejercen un efecto devastador sobre los pacientes y sus familias. En todo el mundo, aproximadamente 300.000 bebés nacen con anemia drepanocítica y 60.000 nacen con β-talasemia cada año. Sin embargo, si los genes de la globina fetal funcional de los pacientes pudieran utilizarse para compensar los genes de la globina adulta mutada, los resultados clínicos podrían mejorarse enormemente. Un reciente estudio proporciona una base para desarrollar dicha terapia.
Hemoglobina fetal
La hemoglobina adulta (HbA) consiste en dos cadenas de α-globina y dos cadenas de β-globina (α2βΑ2). La hemoglobina drepanocítica (HbS, del inglés Sickle hemoglobin) es causada por una sola mutación (Glu6Val) en el gen β-globina (α2βS2). La HbS desoxigenada polimeriza y altera los eritrocitos de tal manera que se alargan, adoptando una forma de hoz frágil e inflexible. Los pacientes con anemia falciforme tienen una variedad de complicaciones agudas y crónicas que incluyen hemólisis y anemia, crisis dolorosas e infarto de los órganos principales. Los pacientes dependientes de transfusiones con talasemia heredan mutaciones en sus genes β-globina que reducen o eliminan la expresión de la globina β, lo que resulta en un desequilibrio en la cadena polipeptídica y en una eritropoyesis ineficaz, que conduce a una anemia potencialmente mortal.
Durante la vida fetal, los genes de γ-globina en lugar de los genes de β-globina están activos. Producen HbF (hemoglobina fetal [α2γ2]) en lugar de HbA. Durante el primer año de la vida posnatal, la expresión del gen γ está disminuida, mientras que lo contrario ocurre con el gen β. En los adultos, el nivel de HbF suele ser de alrededor del 1% de la hemoglobina total. Sin embargo, el nivel de HbF es variable y en algunas personas con enfermedad de células falciformes y talasemia que tienen niveles comparativamente altos niveles de HbF tienen mejores resultados clínicos que aquellos que tienen niveles bajos de HbF. La hidroxiurea, un fármaco que se utiliza con frecuencia para reducir la gravedad de los síntomas de las hemoglobinopatías, puede actuar, en parte, elevando los niveles de HbF. Un fármaco que aumenta más eficazmente la HbF es un santo grial de la investigación de la hemoglobinopatía.
El factor de transcripción BCL11A es un represor crítico de la globina γ en adultos, y los polimorfismos de BCL11A influyen en los niveles de HbF y en la gravedad clínica en pacientes con hemoglobinopatías. Sin embargo, dirigirse directamente a estos factores es difícil. Un enfoque más prometedor podría ser modificar las enzimas que controlan los factores de transcripción. Grevet y colaboradores efectuaron un cribado genético para buscar proteínas quinasas críticas para la regulación de la HbF en una línea celular humana inmortalizada que puede ser diferenciada en células de la línea eritroide (Science 2018;361:285-290).
Pero antes de diferenciar la línea celular, Grevet y sus colegas primero interrumpieron genéticamente cada dominio conocido de la proteína quinasa humana: 496 dominios de quinasa en 482 genes fueron alterados a través de un grupo de células. Las células editadas en este grupo se diferenciaron a eritroblastos y se examinaron para detectar altos niveles de proteína HbF mediante citometría de flujo. Se encontraron altos niveles de HbF en células en las que se interrumpió el inhibidor regulado por el grupo hemo (HRI [también conocido como EIF2AK1]), una quinasa altamente expresada en los eritroblastos (figura 1). Este resultado sugirió que el HRI, que ya ha estado implicado en la coordinación de la síntesis de globina y hemo, reprime la expresión de la HbF en glóbulos rojos humanos.
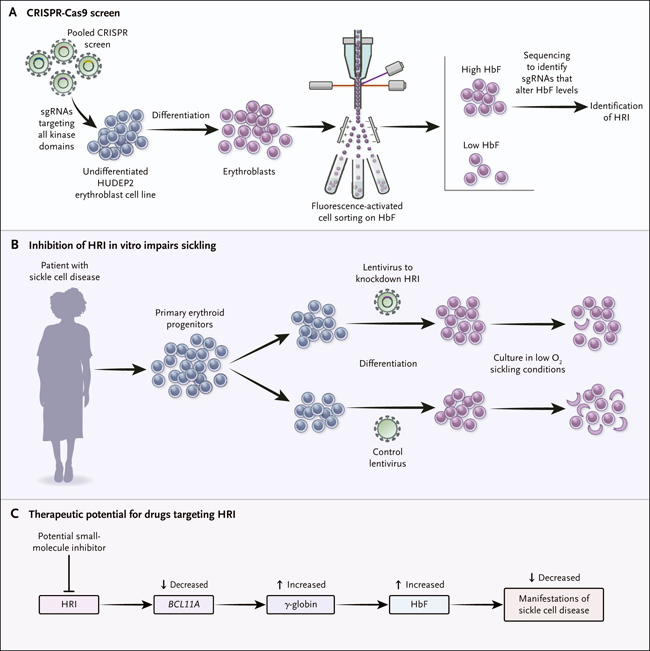
Figura 1. Efecto del inhibidor regulado por hemo en la hemoglobina fetal.
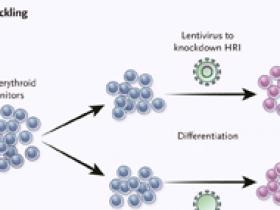
Grevet y sus colegas (Science 2018;361:285-290) realizaron un estudio en el cual se enfocaron sistemáticamente en todas las quinasas conocidas en una línea celular de eritroblastos de humano (panel A). Midieron los niveles de hemoglobina fetal (HbF) con citometría de flujo, y compararon las quinasas dirigidas a las células que expresan niveles altos de HbF con las dirigidas a las células que expresan niveles bajos. El inhibidor regulado por el grupo hemo (HRI) se identificó como la única quinasa que, cuando se suprime a través de la alteración de su gen, causa una alta expresión de HbF. Posteriormente, los autores demostraron que la interrupción de HRI afecta la anemia drepanocítica in vitro (panel B). Las células progenitoras eritroides de una persona con enfermedad de células falciformes se infectaron in vitro con un lentivirus que previene la expresión de la HRI y luego se diferenciaron en glóbulos rojos y se sometieron a condiciones de anemia falciforme por falta de oxígeno. Se observó una disminución de la anemia en los eritrocitos que carecían de HRI. Un inhibidor del HRI podría disminuir la expresión de BCL11A y, por lo tanto, aumentar la producción de globina y HbF (panel C). El aumento de los niveles de HbF podría reducir las manifestaciones de la hemoglobinopatía y atenuar la gravedad de la enfermedad. CRISPR denota repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas, HUDEP2, progenitor eritroide derivado de la sangre del cordón umbilical humano 2, y sgRNA, ARN de guía única.
Utilizando secuenciación de ARN y espectrometría de masas, los autores encontraron que el deterioro de HRI ejercía efectos específicos sobre la expresión del ARN mensajero de la globina γ. Es crucial observar que la diferenciación eritroide y la regulación de otros componentes críticos, como otros genes de la globina, las proteínas de la membrana y las enzimas críticas para la síntesis del hemo, no se vieron perturbadas. La deficiencia de HRI en los eritroblastos humanos primarios también aumentó la transcripción de la globina γ y aumentó la producción de HbF, y nuevamente los efectos del agotamiento de HRI fueron relativamente específicos de γ-globina. Un hallazgo importante es que en los eritroblastos generados a partir de células madre de pacientes con drepanocitosis, el agotamiento de la HRI reguló la expresión de la globina γ y de proteína HbF. Además protegió a las células de la anemia falciforme cuando fueron cultivadas en condiciones de bajo contenido de oxígeno (figura 1), lo que sugiere que la inducción de HbF lograda a través de este enfoque sería clínicamente beneficiosa. Finalmente, el knockdown de HRI redujo la expresión de BCL11A al inhibir la transcripción. Cuando los niveles de proteína BCL11A se incrementaron por la sobreexpresión del gen, los efectos de la disminución de HRI sobre la HbF se invirtieron en gran medida. Por lo tanto, el HRI probablemente actúa controlando la expresión de BCL11A.
La inhibición de HRI es una estrategia prometedora para elevar los niveles de HbF en los pacientes, quizás en combinación con otros inductores de HbF conocidos. Sin embargo, actualmente faltan inhibidores farmacológicos eficaces y específicos de HRI. Además, los datos hasta ahora sólo se derivan de modelos celulares en el laboratorio. Será necesaria una caracterización más exhaustiva antes de que un inhibidor de la HRI pueda ser probado en ensayos clínicos. A pesar de estos problemas, el estudio de Grevet y sus colegas destaca el valor de los enfoques de cribado genético para definir posibles blancos farmacológicos y el valor de los enfoques de la economía para validar la especificidad de estas vías antes del desarrollo farmacéutico. El HRI es ahora una molécula interesante para estudios posteriores de investigadores que buscan encontrar una forma de reducir la carga de las hemoglobinopatías en pacientes de todo el mundo.