Presentación de antígenos por las células B en la esclerosis múltiple
Un estudio reciente de una clase de moléculas HLA en la superficie de las celulas B en personas con esclerosis múltiple reveló que muchas de estas moléculas presentan péptidos propios a las células T.
Hasta hace muy poco, la enfermedad desmielinizante autoinmune esclerosis múltiple se consideraba una patología mediada por células T prototípicas, basado en la analogía con los modelos murinos de autoinmunidad del sistema nervioso. Esta visión cambió radicalmente con el desarrollo de mejores modelos y la reexaminación de la inmunopatología de la enfermedad, ambos revelaron un papel crítico de las células B y el sistema inmunitario humoral. Lo más importante, la demostración de que el agotamiento de las células B con anticuerpos monoclonales antiCD20 es eficaz en el tratamiento de todas las formas de la condición, consolidando esta nueva comprensión de que las células B desempeñan un papel central en la patogénesis.
Ahora se reconoce que los linfocitos B restringidos por clonación, especialmente los de memoria antigénica y los plasmoblastos secretores de anticuerpos, circulan entre el torrente sanguíneo y el sistema nervioso central (SNC) y probablemente se activan en ambos compartimentos.
Estas células B son proinflamatorias y producen "bandas oligoclonales" (que se observan tras la electroforesis en gel), la indicación característica de la presencia de moléculas de inmunoglobulina en el líquido cefalorraquídeo (LCR) que se ha utilizado durante mucho tiempo en el diagnóstico de la esclerosis múltiple. Inicialmente parecía probable que los procesos patológicos mediados por las células B en la esclerosis múltiple se debían a estos anticuerpos derivados del SNC, pero los trabajos posteriores indicaron que la mayoría parecen no ser patógenos. La atención se centró entonces en la presentación de antígenos por parte de las células B a las células T. De hecho, en algunos modelos de laboratorio, la presentación de antígenos por parte de las células B es un requisito obligatorio para la generación de células T patógenas y las manifestaciones clínicas de la enfermedad del SNC.
Tanto los factores genéticos como las influencias ambientales, incluida la infección por el virus de Epstein-Barr, contribuyen a la susceptibilidad a la esclerosis múltiple. La asociación entre la susceptibilidad y el HLA-DR15 está bien establecida, y el locus HLA-DR15, que incluye los genes que codifican dos proteínas heterodiméricas, DR2a y DR2b (Figura 1A), representa la mitad del riesgo genético. (Hay dos genes DRB diferentes en este locus; cada uno codifica una cadena β diferente y cada cadena β puede combinarse con la cadena α para formar un heterodímero).
Las proteínas HLA-D (incluida la HLA-DR15), también conocidas como moléculas del complejo mayor de histocompatibilidad (MHC) de clase II, se expresan principalmente en los linfocitos B y células mieloides y son necesarias para unir y presentar fragmentos de péptidos a las células T CD4+ antígeno específicas.
En conjunto, la asociación entre HLA-DR15 y la esclerosis múltiple, la presencia de células T en las lesiones de la esclerosis múltiple, y la demostración de que los clones de células T CD4+ específicos para la proteína de la mielina pueden causar parálisis recurrentes y desmielinización del SNC en modelos animales, sugieren fuertemente que las células T CD4+ contribuyen a los procesos patológicos de la enfermedad. Sin embargo, la identificación de dianas peptídicas que se unen a HLA-DR15 y activan las células T CD4+ patógenas ha eludido a los investigadores.
En 1995, Wucherpfennig y Strominger demostraron que algunos clones de células T específicos de la proteína de la mielina restringidos por HLA-DR15 de pacientes con esclerosis múltiple también reconocían (además de la mielina) péptidos estructuralmente similares del virus de Epstein-Barr y otros virus, lo que sugiere que estos "imitadores moleculares" podrían desencadenar células T patógenas.
Fue una sorpresa cuando Jelcic y colaboradores observaron en 2018 que las células B de memoria de pacientes HLADR15+ con esclerosis múltiple podían activar las células T CD4+ en ausencia de proteínas exógenas e identificaron un péptido de la proteína RAS liberadora de guanilo 2 (RASGRP2), una proteína intracelular, responsable de esta autoproliferación de las células T. RASGRP2 también se expresa en las neuronas de la materia gris cortical, lo que lleva a la posibilidad de que la presentación de RASGRP2 por las moléculas HLA-DR15 expresadas en las células B estimule a las células T CD4+ que luego se dirigen a RASGRP2 en el cerebro.
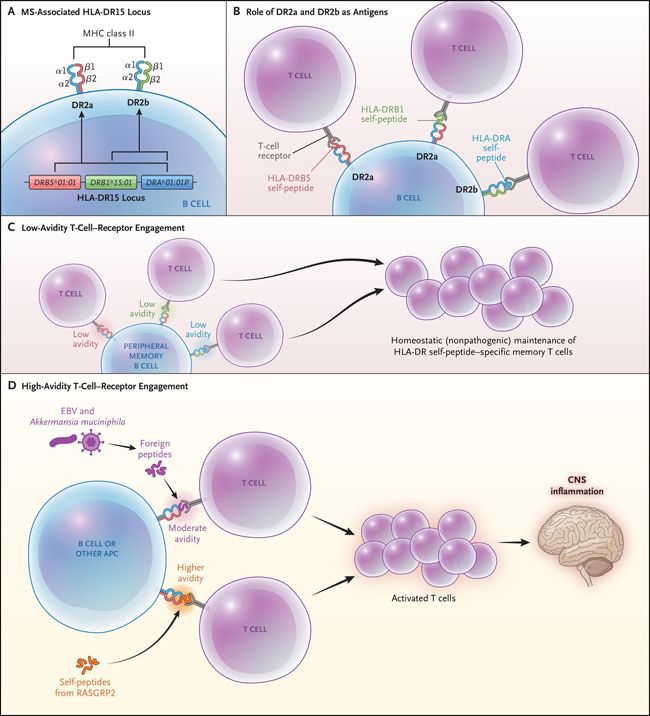
Figura 1: Péptidos propios asociados al HLA-DR15 en las células B y repertorio de células T autorreactivas en la esclerosis múltiple.
El panel A muestra el locus HLA-DR15 asociado a la esclerosis múltiple (EM). Las proteínas HLA-D, que incluyen tres isotipos (HLA-DQ, HLA-DP y HLA-DR), son moléculas de histocompatibilidad (MHC) de clase II compuestas por heterodímeros de cadena α y β. Las moléculas MHC clase II, que se expresan en las células presentadoras de antígenos (CPA) (por ejemplo, las células B, los monocitos y microglía del sistema nervioso central), son necesarias para la presentación de fragmentos peptídicos procesados de antígenos extraños o autoantígenos a las células T CD4+. El haplotipo DR15 contiene un gen de cadena α, DRA*01:01P, y dos genes de cadena β, DRB5*01:01 y DRB1*15:01. El emparejamiento de la cadena α con la β codificada por DRB5*01:01 o la cadena β codificada por DRB1*15:01 crea un heterodímero DR2a o DR2b, respectivamente. Como se muestra en el panel B, un estudio reciente de Wang y colaboradores demostró que los fragmentos de péptidos propios derivados de las proteínas DR2a y DR2b se unen a las moléculas DR2a y DR2b intactas en células B y sirven como antígenos para su presentación a las células T. Los péptidos propios derivados de las cadenas β DRB5 (rojo) y DRB1 (verde) se unen al DR2a, y los péptidos propios de la cadena α, DRA (azul), se unen al DR2b. El panel C muestra que los péptidos propios HLA, que representan una gran proporción de péptidos que se unen a las moléculas DR2a y DR2b en las células B de memoria periféricas en pacientes EM, muestran un compromiso con los receptores de las células T de baja validez, apoyando el mantenimiento homeostático (no patogénico) de células T de memoria específicas de péptidos HLA-DR. El panel D muestra cómo los péptidos pequeños patógenos (por ejemplo, péptidos derivados de la proteína liberadora de guanilo RAS 2 [RASGRP2], que se expresa en las células B, así como por las neuronas de la materia gris cortical) pueden sustituir DR2a y DR2b, lo que conduce a un compromiso de los receptores de las células T de alta validez, lo que promovería la activación de células T CD4+ con péptidos patógenos, que luego migrarían al SNC. Los péptidos extraños derivados de ciertos organismos (por ejemplo, el virus de Epstein-Barr [EBV] y Akkermansia muciniphila) que tienen una avidez moderada también pueden sustituir a los péptidos propios DR2a y DR2b y activar las células T CD4+ que migran al SNC. Las CPA del SNC que expresan DR2a y DR2b pueden presentar el correspondiente péptido patógeno expresado en el SNC (por ejemplo, RASGRP2) a las células T CD4+, provocando respuestas inflamatorias y daños en los tejidos.
En un estudio reciente del grupo de Wang caracterizaron el conjunto de péptidos propios procesados endógenamente que se unen a las moléculas DR2a y DR2b en los linfocitos B y monocitos, una subpoblación mieloide de células presentadoras de antígenos. Aunque los "inmunopéptidos" derivados de las células B y monocitos mostraban cierto solapamiento también había claras diferencias. Más de la mitad de los péptidos propios eluidos de DR2a y DR2b en células B se derivaban de las propias moléculas DR2a y DR2b. Lo más llamativo es que el DR2a presentó preferentemente péptidos propios de las cadenas DRβ (codificados por DRB5*01:01 y DRB1*15:01) y DR2b presentó preferentemente péptidos de la cadena DRα (codificada por DRA*01:01P). Así pues, los DR2a y DR2b expresados por las células B pueden tener doble función en la esclerosis múltiple, ya que actúan como moléculas presentadoras de antígenos y como fuente de epítopos peptídicos.
Las células T CD4+ de memoria derivadas del LCR respondieron a los péptidos propios en asociación con las moléculas DR2a o DR2b y, en algunos casos, también reaccionaron de forma cruzada con la proteína básica de mielina, lo que apoya el mimetismo molecular.
La reactividad cruzada de las células T entre los péptidos propios de HLA-DR y RASGRP2 fue aún más común, y un clon de células T que reconoció a RASGRP2 también reaccionó a virus de Epstein-Barr y Akkermansia muciniphila, una bacteria intestinal comensal asociada a la patogénesis de la esclerosis múltiple. Se observó una jerarquía de antígenos, en la que RASGRP2 era el agonista más fuerte, virus de Epstein-Barr y A. muciniphila eran agonistas moderadamente fuertes, y los péptidos propios HLA-DR eran agonistas débiles. Estos datos sugieren que los péptidos propios unidos al DR2a y al DR2b de las células B contribuyen a conformar el repertorio autorreactivo de las células T CD4+ en la esclerosis múltiple.
Los resultados obtenidos por Wang y colaboradores plantean varias preguntas. Los péptidos propios dominantes unidos a las moléculas HLA-DR de las células B no se detectaron en el tejido cerebral de pacientes con esclerosis múltiple. ¿La ausencia de autopéptidos HLA-DR en las células presentadoras de antígenos del SNC proporciona una mayor oportunidad para que otros péptidos, posiblemente patógenos, se unan a estas moléculas de clase II y activen las células T en el sistema nervioso? ¿Es el RASGRP2, que se expresa en las neuronas corticales, así como en las células B, el tan buscado antígeno objetivo "iniciador" de la esclerosis múltiple? En sus primeras fases, la esclerosis múltiple se considera principalmente una enfermedad desmielinizante, mientras que la pérdida neuronal (neurodegeneración) se vuelve predominante a medida que la enfermedad progresa. Otra posibilidad es que RASGRP2 sea un objetivo secundario del SNC, similar a las proteínas ubicuas intracelulares que presumiblemente se exponen durante el daño inicial y son reconocidas por anticuerpos en el LCR (los que forman las bandas oligoclonales mencionadas anteriormente).
Estos nuevos datos tienen varias implicaciones para entender la inmunidad, autoinmunidad y esclerosis múltiple. El repertorio de antígenos presentados por las células B no se limita a las proteínas reconocidas por su idiotipo de anticuerpos de superficie, como se pensaba anteriormente, sino que incluye rico conjunto de péptidos endógenos. Además, el repertorio de péptidos presentados exclusivamente por células B, y no por otras células presentadoras de antígenos, también es amplio. Es notable que una proporción sustancial de estos péptidos presentados por las moléculas HLA de clase II en las células B parecen ser fragmentos de las propias moléculas de clase II. Además, algunos de estos péptidos propios derivados de las moléculas de clase II son una fuente de mimetismo molecular con autoantígenos virales, bacterianos o de superficie celular, lo que posiblemente desencadena, mantiene o regula la autoinmunidad mediada por células T. Por último, cabe destacar que la arquitectura genómica del gen asociado a la esclerosis múltiple, haplotipo HLA-DRB1*15, es única por la presencia "adicional" del alelo DRB (DRB5*01:01) que está ausente en la mayoría de los demás haplotipos HLA de clase II comunes en humanos. Quizás esta característica ayude a explicar su asociación genética con el riesgo de esclerosis múltiple, ya que puede servir como fuente de péptidos de reacción cruzada con antígenos encefalitogénicos. Las observaciones de Wang y colaboradores seguramente estimularán los esfuerzos por identificar los péptidos unidos al HLA-DR en diferentes clases de células presentadoras de antígenos que participan en la esclerosis múltiple y otras enfermedades autoinmunes, y renueven el interés por avanzar en los enfoques para manipular la expresión de los péptidos asociados al DR con fines terapéuticos.
Fuente bibliográfica
Antigen Presentation by B Cells in Multiple Sclerosis
Scott S. Zamvil, M.D., Ph.D., and Stephen L. Hauser, M.D.
UCSF Weill Institute for Neurosciences, Department of Neurology (S.S.Z., S.L.H.), and the Program in Immunology, University of California, San Francisco (S.S.Z)
N Engl J Med 2021; 384:378-381