Editando el genoma mitocondrial
Para la mayoría de los pacientes con trastornos mitocondriales, muchos de ellos causados por mutaciones en el genoma mitocondrial, solo se dispone de tratamiento sintomático. Un informe reciente publicado describe un estudio de prueba de principio que involucra la edición enzimática del ADN mitocondrial.
Las enfermedades mitocondriales son un heterogéneo grupo de trastornos, que suelen tener características clínicas variables generadas por una función alterada de la cadena respiratoria mitocondrial. La subyacente causa serían mutaciones genéticas que afecten al ADN nuclear o al ADN mitocondrial (ADNmt), los cuales contienen genes que codifican componentes de la maquinaria de fosforilación oxidativa necesaria para generar ATP, clave de la energía celular. Para la mayoría de los pacientes, solo se dispone de tratamiento sintomático y se necesitan nuevas terapias dirigidas directamente a los mecanismos involucrados. Un espectro de más de 250 enfermedades mitocondriales puede vincularse con una variedad de variantes patogénicas que ocurren en los genomas nucleares o mitocondriales; estas patologías se manifiestan típicamente como trastornos multisistémicos graves y a menudo letales. Aunque cada una es muy rara, en conjunto, las enfermedades mitocondriales afectan de 5 a 15 personas por cada 100.000 habitantes. Las mutaciones patogénicas en el ADNmt son más frecuentes en adultos que las mutaciones en genes nucleares. Se han informado de unas 270 variantes patogénicas de ADNmt.
Las características clínicas y la herencia de los trastornos por mutación del ADNmt se diferencian de los trastornos autosómicos. Un concepto clave es la heteroplasmia: coexistencia de variantes normales y patógenas dentro de las células afectadas por enfermedades del ADNmt. En una célula normal, el pequeño genoma mitocondrial circular está presente en miles de copias de ADNmt, y el número de genomas mutantes debe alcanzar un nivel crítico de heteroplasmia, o umbral, para causar disfunción celular y enfermedad mitocondrial (figura 1A). En investigaciones anteriores, se han realizado esfuerzos para desarrollar terapias que eliminen específicamente las variantes patógenas del ADNmt hasta que su número esté por debajo de los niveles de umbral. Con este fin, las nucleasas de ADN, como las efectoras de tipo activador de la transcripción (TALEN) y las nucleasas dedos de zinc, se han diseñado genéticamente para apuntar a copias mutantes del ADNmt. Después de la linealización, las copias de ADNmt mutante se degradan, observándose un cambio descendente en los niveles de heteroplasmia, tanto en modelos celulares como de ratones. Un enfoque alternativo sería corregir o editar específicamente los pares de bases mutados. Recientemente, las tecnologías CRISPR-Cas (repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas asociadas con una endonucleasa Cas) han revolucionado la capacidad de cambiar el genoma nuclear a voluntad. Sin embargo, se requiere un ARN guía y, dado que las cadenas largas de ADN y ARN no se pueden importar a las mitocondrias de mamíferos, la tecnología no se utiliza para editar el ADNmt.
Sin embargo, en una publicación reciente, Beverly Y. Mok y colaboradores describen nuevas herramientas para la manipulación precisa de secuencias de ADNmt. Con este fin, los autores utilizaron creativamente una citidina desaminasa para convertir de manera eficiente sitios específicos en el ADNmt. La clave de esta técnica fue la identificación de una citidina desaminasa de Burkholderia cenocepacia, la DddA. Esta enzima funciona como una toxina interbacteriana que desamina la citosina (C) precedida por un residuo de timina (T) en el ADN bicatenario. Los autores dividieron el dominio DddA que causa toxicidad (DddAtox) en dos mitades inactivas, que estaban vinculadas a proteínas programables de unión al ADN. Cuando esas proteínas de fusión se unen a sitios diferentes pero adyacentes en el ADNmt, las mitades de DddAtox divididas se juntan y la desaminasa se activa (figura 1B). Las proteínas de unión al ADN se programan para dirigirse a sitios de modo que el DddAtox reactivado se extienda a ambos lados del ADNmt de doble hebra en la C diana (precedida por T). La desaminación convierte a la C en uracilo (U), una nucleobase que normalmente se empareja con la adenina (A). Como consecuencia, cuando el ADN editado se usa como molde para la replicación del ADNmt, se introduce A frente al U. De esta manera, el par de bases CG original se convierte en un par de bases UA después de una ronda de replicación del ADNmt. Finalmente, las vías de reparación celular reemplazan el U por T, formando TA y completando la edición de la base. Los autores demostraron la eficiencia de este sistema en líneas celulares, donde se podría convertir CG en TA en secuencias diana específicas en 5 a 50% de las secuencias de ADNmt probadas.
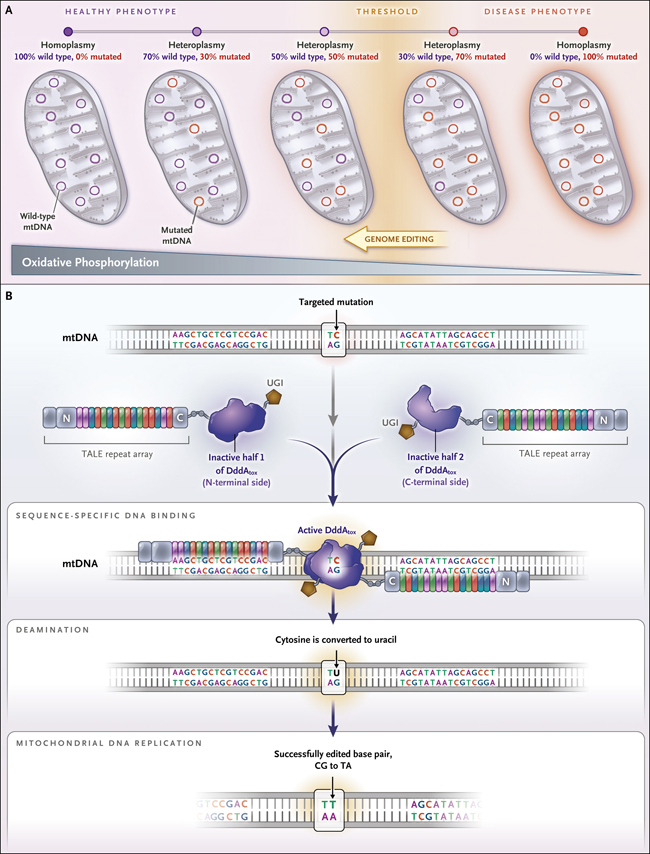
Figura 1: nuevas herramientas para editar el genoma mitocondrial
En la mayoría de las mutaciones causantes de enfermedades en el ADN mitocondrial (ADNmt), el nivel y la distribución tisular de la heteroplasmia determina las manifestaciones fenotípicas (panel A). Una célula humana típica contiene más de 1000 copias de ADNmt, y la presencia de mutaciones en una fracción sustancial tiene pocos o ningún efecto nocivo. Una menor actividad de fosforilación oxidativa y los fenotipos de enfermedades no suelen observarse hasta que el nivel de ADNmt mutado excede el umbral específico. La edición genómica corrige las mutaciones del ADNmt y, por tanto, modificar los niveles de heteroplasmia por debajo del umbral requerido para que ocurran los fenotipos de la enfermedad. Al usar un editor base derivado de una toxina interbacteriana llamada DddA (editor de base de citosina derivada de DddA [DdCBE]), se edita el ADNmt directamente (panel B). El dominio de citidina desaminasa de la toxina (DddAtox) se divide en dos partes inactivas, cada una fusionada a un dominio de unión al ADN específico de secuencia diseñado (un efector similar al activador de la transcripción [TALE]). Cuando las construcciones TALE se unen a secuencias de ADN adyacentes, las mitades de DddAtox se alinean, formando una desaminasa activa. La desaminación de C a U en el ADNmt conduce a la formación de un par de bases UG. Los inhibidores de uracilglicosilasa (UGI) evitan que la maquinaria de reparación del ADNmt elimine el U. Durante la siguiente ronda de replicación del ADNmt, el U se une con A, lo que eventualmente conduce a la conversión de un CG en un par de bases TA.
El editor de bases de citosina derivada de DddA (DdCBE) permite la edición directa de ADNmt. Esta herramienta también se puede utilizar para generar mutaciones de ADNmt en líneas celulares y potencialmente en modelos animales. Clínicamente, la técnica promete editar los pares de bases de ADNmt de CG mutante, corrigiendo casi la mitad de las variantes de ADNmt patógenas conocidas. Sin embargo, la versión actual de DdCBE solo puede editar citosinas precedidas por timina, y los sistemas de administración de genes deberán superar los desafíos de la administración generalizada de dos DdCBE en los tejidos para tratar enfermedades del ADNmt, que son multisistémicas. Además, la eficiente edición de bases requiere una replicación activa del ADNmt, y muchas mutaciones patológicas del mtDNA causan fenotipos en tejidos posmitóticos, como el cerebro y el músculo. No está claro si los bajos niveles de replicación del ADNmt que tienen lugar continuamente en estas células son suficientes para una eficiente edición de base. Seguramente se perfeccionará la técnica, que conduzca a una mejor comprensión de las variantes del ADNmt en enfermedades y afecciones relacionadas con el ADNmt humano tales como el envejecimiento, sin mencionar el tratamiento o la prevención de los trastornos primarios por mutación del ADNmt.
Fuente bibliográfica
Editing the Mitochondrial Genome
Maria Falkenberg, Ph.D., and Michio Hirano, M.D.
Institute of Biomedicine, University of Gothenburg, Gothenburg, Sweden (M.F.), and the H. Houston Merritt Neuromuscular Research Center, Columbia University Medical Center, New York (M.H.).
N Engl J Med 2020; 383:1489-1491