Genética Cínica
Perfil epigenético para la detección de tumores

Mediante análisis de sangre se logra identificar células cancerígenas y establecer su origen basado en patrones de metilación del ADN.
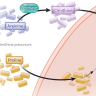
La metilación de sitios CpG en mamíferos es una modificación epigenética relativamente estable, que puede transmitirse a través de la división celular. Tal cambio es específico para cada tipo de célula y puede ser aprovechado para analizar la composición celular relativa de muestras heterogéneas, tales como glóbulos blancos en sangre, componentes fetales en el ADN circulante materno libre de células (cfDNA, por sus siglas en inglés) o ADN tumoral circulante (ctDNA) en plasma. Específicamente, los sitios CpG adyacentes de los genomas de mamíferos pueden co-metilarse debido a la procesividad de enzimas metiltransferasas, pero también se han observado patrones de metilación discordantes, que están relacionados con procesos moleculares estocásticos o no coordinados, particularmente en el cáncer.
Shicheng Guo y especialistas de la Universidad de California en San Diego se centraron en una búsqueda sistemática e investigación de regiones del genoma humano completo que muestran metilación altamente coordinada. Se definieron 147.888 bloques de sitios CpG fuertemente acoplados, denominados bloques de haplotipos de metilación, después del análisis de 61 conjuntos de datos de secuenciación de todo el genoma. Usando la métrica de carga de haplotipo de metilación, se realizó el análisis de tejido específico a nivel de bloque. Finalmente, se identificaron subconjuntos de bloques informativos para muestras heterogéneas.
Con la información obtenida, se desarrolló un modelo predictivo para estimar la presencia y el origen de un tumor a partir de ADN libre circulante en sangre. Este modelo combina dos señales diferentes: una que detecta el ADN de las células tumorales y otra que permite estimar el tejido de origen. Por último, se aplicó el modelo en muestras de sangre de 59 pacientes con cáncer de pulmón o colon. En este caso, el análisis de ADN circulante en plasma permitió detectar no sólo el ADN circulante anómalo sino también el material liberado por las células normales del tejido afectado, las que mueren durante el proceso tumoral.
Finalmente, utilizando haplotipos de metilación se muestra una estimación cuantitativa de la carga tumoral y el tejido de origen del ADN libre de células de 59 pacientes con cáncer de pulmón o colorrectal.
Temas Relacionados