Mutación neuroprotectora
Los portadores de mutaciones causantes de la enfermedad de Alzheimer que permanecen sin impedimentos cognitivos hasta edades más avanzadas podrían ayudar al descubrimiento de genes que reducen el riesgo. Recientemente, se identificó un portador de la mutación PSEN1 (presenilina 1) asociado con la enfermedad de Alzheimer autosómica dominante, que no desarrolló un deterioro cognitivo leve hasta los setenta años, tres décadas después de la edad esperada de su inicio clínico. Esta persona, tenía dos copias de la mutación APOE3 Christchurch (R136S), niveles inusualmente altos de amiloide cerebral y mediciones limitadas de tau y neurodegeneración. Estos reveladores hallazgos tienen implicaciones para el papel de la APOE en la patogénesis, tratamiento y prevención de la enfermedad de Alzheimer.
Mutación APOE3-R136S
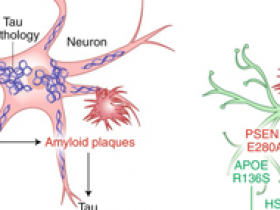
Actualmente se piensa que la enfermedad de Alzheimer (EA) es causada por complejas interacciones entre múltiples factores genéticos, epigenéticos y ambientales. Las mutaciones en tres genes -APP (precursor de la proteína amiloide), PSEN1 (que codifica la presenilina 1) y PSEN2- causan la EA autosómica dominante de inicio temprano (ADAD). PSEN1 y PSEN2 procesan proteolíticamente a la APP para generar varios péptidos beta amiloide (Aβ) y otros productos proteicos en el cerebro. Las mutaciones en los tres genes afectan al procesamiento de APP, alterando la producción de los diferentes péptidos Aβ y, por tanto, sus concentraciones relativas y su propensión a la agregación. Juntos, estos cambios conducen a un aumento de la formación de placa amiloide en los cerebros de los individuos con ADAD. Se ha sugerido ampliamente que los niveles elevados de agregados Aβ, o de placas amiloides conducen a patologías de la proteína tau y posteriormente a un declive cognitivo relacionado con la EA (figura 1, izquierda). La apolipoproteína E, cuya función principal es el transporte de lípidos en los tejidos periféricos y en el cerebro, tiene tres isoformas comunes: APOE2, APOE3 y APOE4. La APOE4 está vinculada con la EA familiar y esporádica de inicio tardío; su dependencia de la dosis genética aumenta el riesgo de desarrollar EA y disminuye la edad de inicio de la enfermedad en los portadores. También se ha informado de que aumenta la acumulación de Aβ y exacerba la patología tau (figura 1, izquierda).
La familia de individuos con ADAD más grande conocida en el mundo, los portadores de la mutación colombiana PSEN1-E280A, comprende más de 1.200 pacientes. Al igual que otras mutaciones de PSEN1, la mutación PSEN1-E280A aumenta la producción de Aβ, lo que conduce a la acumulación de amiloide en el cerebro. En este sentido, aunque existe cierta variabilidad en la edad de inicio y en la progresión de la enfermedad, tanto los hombres como las mujeres portadoras de la mutación tienen un curso de la enfermedad notablemente rápido y consistente, desarrollando un deterioro cognitivo leve y demencia a las edades medias de 44 (IC 95%, 43-45) y 49 (IC 95%, 49-50) años, respectivamente. La identificación de variantes genéticas capaces de proteger a los portadores de la mutación PSEN1-E280A del desarrollo de la EA de inicio precoz es de fundamental importancia para comprender mejor la patogénesis de la EA y para desarrollar estrategias para tratar o prevenir el EA.
En un estudio reciente, Arboleda-Velasquez y colaboradores informan sobre la identificación de una mutación homocigótica de APOE, APOE3-Christchurch (R136S), que confiere una fuerte protección contra el desarrollo de la ADAD en una portadora de PSEN1-E280A (figura 1, derecha), abriendo una nueva y prometedora vía en la investigación y terapéutica de la EA.
Los autores identificaron a una portadora de PSEN1-E280A que no desarrolló un deterioro cognitivo leve hasta los setenta años, tres décadas después de la edad esperada del inicio clínico. También presentó dos copias de la mutación R136S, extremadamente rara, en APOE3, que los autores sugieren que es responsable de su resistencia a la mutación familiar PSEN1, altamente penetrante de ADAD. Los autores encontraron que esta persona tenía una carga amiloide cerebral inusualmente alta en comparación con otros portadores de PSEN1-E280A, pero solo cantidades limitadas de ovillos neurofibrilares de tau u otros signos neurodegenerativos detectables por imagenología cerebral, incluyendo firmas asociadas a la EA de hipometabolismo cerebral y atrofia del hipocampo (figura 1, derecha). Así, este caso ilustra una clara disociación de Aβ/acumulación amiloide de la patología tau, la neurodegeneración y el declive cognitivo temprano típico de los portadores de mutaciones PSEN1 causantes de la EA.
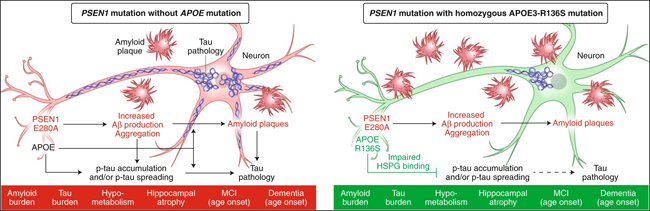
Figura. 1: Efectos de APOE3-R136S en Aβ y patologías tau, neurodegeneración y cognición en portadores de la mutación PSEN1, con posibles mecanismos subyacentes.
A la izquierda, resumen de la imagenología cerebral y las evaluaciones clínicas de los pacientes jóvenes con deterioro cognitivo leve (MCI) y mutación PSEN1 y los roles interactivos ampliamente discutidos de Aβ, APOE y tau en la patogénesis de la EA. A la derecha, resumen de imagenología cerebral y evaluaciones clínicas de un paciente anciano con MCI, mutación PSEN1 y una mutación homocigótica APOE3-R136S, con los mecanismos subyacentes potenciales de los efectos protectores de la EA de APOE3-R136S sobre la ADAD. p-tau, tau fosforilada.
Como se ha mencionado anteriormente, una teoría dominante en la investigación de la EA ha sido que el aumento de los niveles de Aβ, los agregados de Aβ o las placas amiloides conducen a patologías asociadas con tau y posteriormente a un declive cognitivo relacionado con la EA (Figura 1, izquierda). Claramente, los nuevos hallazgos no apoyan esta hipótesis tan extendida. De hecho, el estudio proporciona pruebas directas de que Aβ/acumulación amiloide por sí sola no es suficiente para causar EA, al menos en un portador de la mutación PSEN1 con una carga de placa Aβ excesivamente alta. Alternativamente, Aβ podría inducir patologías tau y declive cognitivo sólo en presencia de APOE normalmente funcional. Un mayor entendimiento de esta cadena de causalidad será vital para comprender mejor la patogénesis de la EA y mejorar el desarrollo de medicamentos.
Los nuevos hallazgos indican que el efecto protector de la mutación APOE3-R136S puede realizarse a través de mecanismos que limitan la patología y la neurodegeneración por tau en presencia de una alta acumulación de Aβ/amiloide (figura 1, derecha). Los autores señalan que la mutación APOE3-R136S afecta a una región de la APOE que se sabe desempeña un papel clave en la unión a los receptores de lipoproteínas, como el receptor de lipoproteínas de baja densidad (LDLR), y a los proteoglicanos de heparán sulfato (HSPG). Es importante destacar que los HSPG promueven la captación neuronal de tau extracelular, lo que puede exacerbar la diseminación de tau y las patologías. El propio análisis de los autores demostró que la APOE3-R136S tiene una afinidad de unión a la heparina marcadamente menor que cualquiera de las tres isoformas comunes de la APOE, incluida la APOE2. Ampliando estas observaciones, los autores generaron un anticuerpo contra la región R136S de la APOE y descubrieron que podía tanto unirse con éxito a la APOE3 como reducir su capacidad de unión a la heparina. A partir de estos datos, los autores plantean la hipótesis de que los anticuerpos u otras moléculas que modulan la interacción APOE-HSPG podrían reproducir el efecto protector contra la EA de APOE3-R136S, incluyendo su potencial para inhibir la diseminación de tau (figura 1, derecha). Esta teoría justifica la realización de pruebas experimentales en células y animales y, de tener éxito, la realización de estudios clínicos adicionales en humanos.
Este estudio da pasos importantes para abordar varias cuestiones que se han debatido en el campo de la APOE y la EA durante décadas. En el cuarto de siglo transcurrido desde que la mutación C112R de la APOE1,5, que da origen a la isoforma APOE4, fue identificada por primera vez como el principal factor de riesgo genético de la EA, su penetración incompleta ha llevado a muchos a considerarla como un modificador en lugar de un factor causal de la EA. Este nuevo estudio demuestra, por primera vez, que la APOE funcional es realmente necesaria para el desarrollo patológico y clínico completo de la EA, al menos en la EA con una carga masiva de placa amiloide. Además, se ha debatido si la APOE4 y la isoforma protectora de la EA, la APOE2, modulan la patogénesis de la EA a través de los efectos de pérdida de función o de ganancia de toxicidad. Este estudio sugiere que la pérdida de la función de unión al receptor o HSPG de la APOE3-R136S, que también se observa en diversos grados con la APOE2, es protectora de la EA, lo que apoya indirectamente la probabilidad de que la APOE4 tenga un efecto de ganancia de toxicidad en la patogénesis de la EA. Finalmente, este estudio apoya la idea de que disminuir en vez de aumentar el nivel de APOE podría ser una estrategia efectiva para tratar o prevenir la EA, independientemente de la isoforma APOE
Sin embargo, se debe tener cierta precaución al interpretar y generalizar los hallazgos de este estudio, ya que solo reporta un caso de la mutación homocigótica APOE3-R136S. Se necesitan más casos clínicos y estudios experimentales en animales para confirmar estas observaciones y explorar los mecanismos moleculares y celulares subyacentes. Además, aunque la mutación homocigótica APOE-R136S es un fuerte candidato para la causa de la resistencia a la EA observada en este caso, se requiere trabajo adicional para confirmar que esta mutación homocigótica es realmente necesaria para el efecto protector de la EA. Dado que sólo cuatro heterocigotos APOE3-R136S que también son portadores de la mutación PSEN1-E280A han sido identificados en la familia colombiana, se necesitan estudios epidemiológicos que incluyan más individuos heterocigotos para APOE3-R136S y estudios experimentales en células y animales para aclarar aún más el efecto de la mutación heterocigota en la EA.
Además de su clara promesa de abordar la ADAD, este estudio plantea la importante pregunta de si la mutación R136S conferiría un efecto protector contra la AD de inicio tardío relacionada con APOE4. La APOE4, el mayor factor de riesgo genético para esta forma mucho más común de EA, ha demostrado tener tanto efectos de ganancia de toxicidad como una mayor afinidad de unión a la heparina en comparación con las otras isoformas APOE4, lo que sugiere que la introducción de la mutación R136S podría proteger contra los efectos patogénicos de APOE4. Debido a que la mutación APOE-R136S es extremadamente rara en los seres humanos, la generación de modelos de ratones y líneas de células madre pluripotentes inducidas con la mutación en un fondo APOE4, APOE3 o APOE2 mediante la edición de genes sería un paso próximo razonable para capitalizar este emocionante hallazgo para estudios más detallados relacionados con la protección contra la EA de inicio tardío y el desarrollo de medicamentos.
Este importante estudio, que describe un caso notable resistencia a la ADAD altamente penetrante, tiene implicaciones cruciales para el papel de la APOE en la patogénesis y el tratamiento de la EA. Este trabajo abre nuevas y emocionantes vías en la investigación de la enfermedad de Alzheimer y revela posibilidades prometedoras de potenciales terapias.
Fuente bibliográfica
An Alzheimer’s-disease-protective APOE mutation
Kelly A. Zalocusky, Maxine R. Nelson & Yadong Huang
Gladstone Institute of Neurological Disease, San Francisco, CA, USA
Nat Med 25, 1648–1649 (2019)