Descubrimientos en insuficiencia cardíaca
La insuficiencia cardíaca con fracción de eyección preservada (ICFEp) es un síndrome común con alta morbilidad y mortalidad para el que no existen terapias basadas en la evidencia. Recientemente, se ha reportado que el estrés metabólico e hipertensivo concomitante, provocado por una combinación de una dieta rica en grasas y la inhibición de la óxido nítrico sintasa constitutiva en ratones, recapitula las numerosas características sistémicas y cardiovasculares de la ICFEp en humanos. La expresión de uno de los efectores de la respuesta a proteínas desplegadas, la forma de empalme alternativo de la proteína 1 de unión X-box (XBP1), se es baja en el miocardio del modelo de roedor y en pacientes con ICFEp. Mecanísticamente, la disminución de las XBP1 resulta del aumento de la actividad de la óxido nítrico sintasa inducible (iNOS), lo que culmina en un empalme defectuoso de XBP1. La supresión farmacológica o genética de la iNOS, o la sobreexpresión de XBP1 restringida por los cardiomiocitos, mejora el fenotipo de la ICFEp. Por lo tanto, la desregulación impulsada por la iNOS de la vía mediada por XBP1 es un mecanismo crucial de la disfunción cardiomiocítica en la ICFEp.
Respuesta a proteínas desplegadas
En el último decenio, la insuficiencia cardíaca con fracción de eyección preservadaa (en la que la fracción de eyección supera el 50%) se ha hecho cada vez más frecuente y representa 56% de todos los casos de insuficiencia cardíaca. Debido a su creciente prevalencia y persistencia fenotípica y a la ausencia de terapias eficaces, la afección tiene la mayor necesidad insatisfecha de tratamiento en la cardiología moderna.
En la insuficiencia cardíaca con fracción de eyección preservada (en adelante, ICFEp), la alta rigidez diastólica del ventrículo izquierdo es de suma importancia porque provoca un aumento brusco de las presiones de llenado del ventrículo izquierdo durante el ejercicio, la congestión pulmonar y, por lo tanto, la intolerancia al esfuerzo. Un modelo de alta rigidez ventricular izquierda diastólica en la ICFEp sostiene que las afecciones coexistentes, especialmente las metabólicas, inducen la inflamación microvascular coronaria como parte de una respuesta inflamatoria sistémica (J Am Coll Cardiol. 2013 Jul 23;62(4):263-71). Se presume que esta inflamación aumenta la rigidez ventricular izquierda diastólica al aumentar la deposición de colágeno en el intersticio miocárdico y reducir la elasticidad de titina (proteína miofilamentosa larga y distensible que controla la elasticidad de los cardiomiocitos). Recientemente, Gabriele Schiattarella y colaboradores (DOI: 10.1038/s41586-019-1100-z) revelaron recientemente un tercer mecanismo mediante el cual la inflamación microvascular coronaria produce una elevada rigidez diastólica del ventrículo izquierdo en la ICFEp: la supresión de la respuesta a proteínas desplegadas en los cardiomiocitos debido a la expresión de la óxido nítrico sintasa inducible. La supresión de esta respuesta dificulta la degradación celular adecuada de las proteínas desestabilizadas y puede dar lugar a su acumulación intersticial, como ocurre en la amiloidosis de la transtiretina, una causa bien establecida de ICFEp (figura 1).
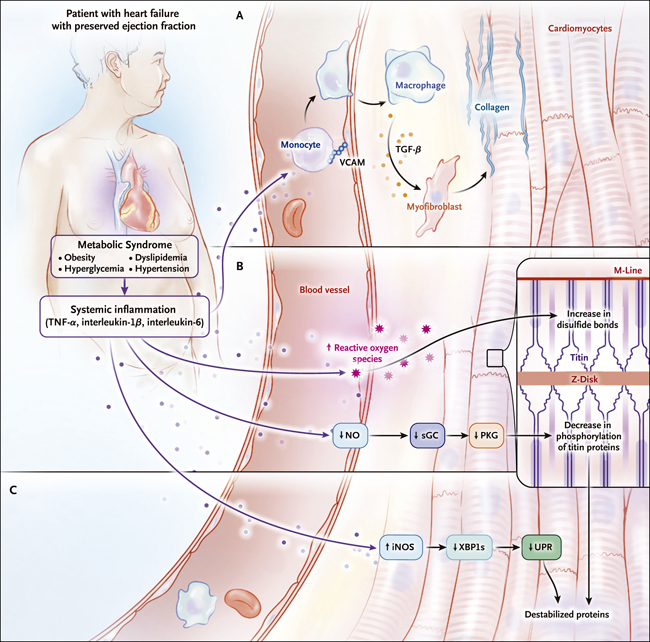
Figura 1. Rutas de modelado desde la inflamación sistémica hasta la rigidez diastólica del ventrículo izquierdo.
La deposición de colágeno, un evento clave en la fibrosis, es una ruta por la cual la inflamación sistémica conduce a la rigidez ventricular izquierda diastólica (panel A). La inflamación sistémica desencadena la expresión de las moléculas de adhesión de las células vasculares (VCAM). Estas moléculas enganchan monocitos que se convierten en macrófagos que secretan el factor de crecimiento transformante β (TGF-β), que estimula a los miofibroblastos a depositar colágeno (panel A). La rigidificación de titina es otra vía por la cual la inflamación sistémica conduce a la rigidez diastólica del ventrículo izquierdo (Panel B). Un estudio de la insuficiencia cardíaca con fracción de eyección conservada en ratas y el análisis de biopsias miocárdicas de muestras obtenidas de personas con esta afección apoyan la opinión de que la inflamación sistémica, caracterizada por la presencia del factor de necrosis tumoral α (TNF-α), la interleucina-1β, y la interleucina-6, está asociada con una menor producción endotelial de óxido nítrico (NO) y una menor actividad de la guanilato ciclasa soluble (sGC) y proteína quinasa G (PKG) en los cardiomiocitos , reduciendo así la fosforilación de titina. La inflamación sistémica también causa la producción de especies reactivas de oxígeno, lo que lleva a la formación de enlaces de disulfuro dentro de titina. Tanto la hipofosforilación como la formación de estos enlaces rigidizan la titina. Schiattarella y sus colaboradores describieron recientemente una tercera vía para la inflamación sistémica: una respuesta deficiente a proteínas desplegadas (UPR). La inflamación sistémica aumenta la expresión de la óxido nítrico sintasa inducible (iNOS), lo que a su vez reduce los niveles de la variante de splicing de la proteína de unión a X-box 1 (XBP1s) y suprime la expresión de las proteínas que ejecutan la respuesta a proteínas desplegadas, lo que puede dar lugar a una acumulación de proteínas desestabilizadas que es similar a la acumulación de transtiretina en la amiloidosis.
¿Cómo se produce la fibrosis y cómo está involucrada la titina? La inflamación microvascular va acompañada de una mayor expresión de moléculas de adhesión en la superficie luminal de las células. Estas moléculas se acoplan a monocitos circulantes, que luego se diferencian en macrófagos al infiltrarse en la microvasculatura y secretar el factor de crecimiento transformante β (TGFβ). TGFβ convierte los fibroblastos en miofibroblastos que depositan colágeno de alta resistencia a la tracción, como el que se encuentra en el tejido cicatricial. La inflamación microvascular también desacopla la óxido nítrico sintasa endotelial, lo que da lugar a niveles más bajos de óxido nítrico y a niveles más altos de especies de oxígeno reactivo, que son tóxicos (figura 2). En los cardiomiocitos subyacentes, los niveles más bajos de óxido nítrico conducen en última instancia a una disminución de la fosforilación de titina y de la elasticidad de los cardiomiocitos.
Schiattarella y colaboradores demostraron que los niveles plasmáticos elevados de citoquinas proinflamatorias potencian la expresión de la óxido nítrico sintasa inducible en los cardiomiocitos. Los niveles elevados de esta enzima dan lugar a una reducción de la actividad de dos proteínas que controlan la respuesta a proteínas desplegadas: una isoforma de la proteína de unión a X-box 1 (XBP1) y la enzima que requiere inositol 1α (IRE1α). Esta última empalma al ARN mensajero XBP1 para producir XBP1s. Queda por demostrar si la supresión de la respuesta a proteínas desplegadas en el cardiomiocito da lugar a una acumulación miocárdica de proteínas desestabilizadas. Las pruebas que respaldan esa acumulación son la elevación de la troponina plasmática en pacientes con ICFEp, que tiene más probabilidades de resultar de la acumulación de proteínas miofilamentosas desestabilizadas que de la muerte celular del cardiomiocito (esta última no se ha observado en las muestras de biopsia del miocardio obtenidas de pacientes con ICFEp).
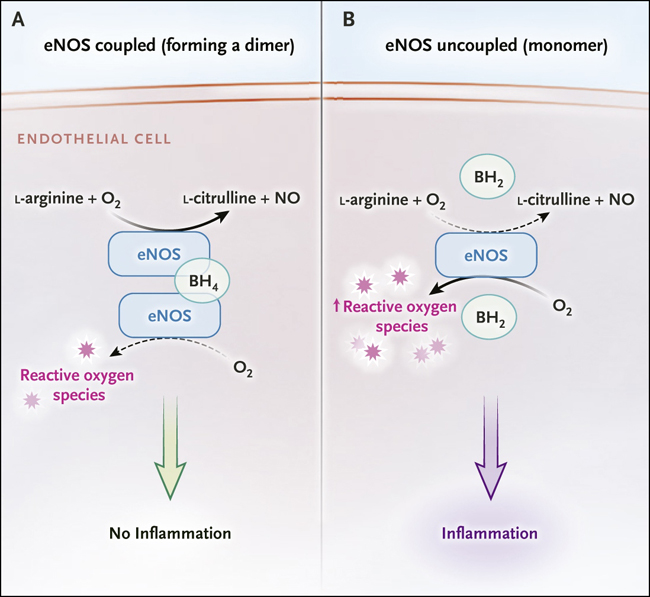
Figura 2. Efecto de la inflamación en la óxido nítrico sintasa.
En condiciones fisiológicas, las moléculas de óxido nítrico sintasa (eNOS) se acoplan a través del cofactor tetrahidrobiopterina (BH4), formando dímeros, y se favorece la vía que produce el óxido nítrico (NO) (panel A). Durante la inflamación, la dihidrobiopterina BH4 no se recicla de BH2, y los dímeros de eNOS se desacoplan para formar monómeros (panel B). La eNOS monomérica favorece una vía alternativa que produce especies reactivas de oxígeno, en lugar de convertir la l-arginina y el oxígeno en l-citrulina y NO. Los cardiomiocitos subyacentes pierden su elasticidad porque los niveles más bajos de NO dan lugar a una reducción de la actividad de la proteína quinasa G y, por tanto, a una reducción de la fosforilación de titina. La elevación resultante en los niveles de las especies reactivas de oxígeno causa entonces la formación de enlaces disulfuros rigidizantes dentro de titina.
Esta forma de insuficiencia cardíaca ha sido modelada en ratas y en animales grandes como perros mayores con hipertensión y cerdos con múltiples condiciones. Sin embargo, Schiattarella y sus colegas, usaron un modelo de ratón con dos "golpes". El primer éxito fue una dieta alta en grasas (que conduce a un compromiso metabólico) y el segundo fue la administración a largo plazo del éster metílico de Nω-nitro-l-arginina, que mediante su potente inhibición de la óxido nítrico sintasa endotelial causa hipertensión arterial. Este nuevo modelo de ratón manifiesta de forma duradera muchas de las características específicas de la insuficiencia cardíaca clínica, como el aumento de peso con intolerancia a la glucosa y una fracción de eyección del ventrículo izquierdo (FEVI) superior al 50% durante un período de hasta 50 semanas. La importancia del compromiso metabólico en el desarrollo de la ICFEp se ilustró comparando los ratones de este nuevo modelo con los ratones que tienen constricción aórtica transversal, en los que se desarrollaron hallazgos "opuestos", entre ellos un nivel elevado de expresión de XBP1 en los cardiomiocitos y una disminución sustancial de la FEVI, que se hizo evidente después de sólo tres semanas. Ambas observaciones apoyan la hipótesis de que la ICFEp es impulsada por la inflamación sistémica resultante de las condiciones metabólicas coexistentes y no por la sobrecarga mecánica. La ventaja de modelar la enfermedad en ratones es que es relativamente fácil probar la prueba de concepto a través de la manipulación genética.
En consecuencia, Schiattarella y sus colaboradores suprimieron genéticamente la óxido nítrico sintasa inducible y sobreexpresaron XBP1 en los ratones afectados. Cada intervención aminoró el fenotipo de la ICFEp: los ratones tratados tenían presiones de llenado del ventrículo izquierdo inferiores y un peso pulmonar menor y podían correr una distancia mayor que los controles. Además, la supresión de la óxido nítrico sintasa inducible confirmó que su supresión disminuyó la actividad de la enzima que requiere inositol 1α a través de la S-nitrosilación, perjudicando su capacidad de empalmar el ARN mensajero de XBP1 para generar la correspondiente proteína.
Estos nuevos conocimientos sobre los mecanismos fisiopatológicos de la alta rigidez diastólica del ventrículo izquierdo apoyan el restablecimiento de los objetivos terapéuticos en pacientes con insuficiencia cardíaca con fracción de eyección conservada. Debe explorarse un enfoque estratificado, con la alineación de la terapia a los mecanismos predominantes. Cuando la fibrosis miocárdica está presente, la terapia antifibrótica podría ser efectiva. En ausencia de fibrosis miocárdica, puede ser apropiado el tratamiento antiinflamatorio o la inhibición de la óxido nítrico sintasa inducible. Por último, si se confirma la acumulación intersticial de proteínas desestabilizadas, la estabilización de las proteínas o la inhibición de la síntesis representarían estrategias experimentales para el tratamiento.
Fuente bibliográfica
Unfolding Discoveries in Heart Failure
Walter J. Paulus, M.D., Ph.D.
Amsterdam Cardiovascular Sciences, Amsterdam University Medical Centers, Amsterdam.
N Engl J Med 2020; 382:679-682