Biología celular
Prionopatías: cuando el beso de la muerte fracasa
Con el paso del tiempo, las proteínas padecen las mismas consecuencias de nuestro propio envejeciendo, es decir, sufren daños en sus estructuras, se pliegan incorrectamente y pierden actividad. Para combatir estos problemas, las células poseen una serie de sistemas intracelulares que reconocen y degradan dichas proteínas. Así, para las proteínas de vida corta, implicadas en numerosos procesos celulares (regulación del crecimiento celular, reparación del ADN, oncogénesis, biogénesis de los ribosomas, infección vírica, degeneración neural y muscular, diferenciación celular, respuesta al estrés, modulación de los receptores de superficie y canales iónicos, procesamiento y presentación de antígenos y activación de factores de transcripción), interviene la vía de la ubiquitina-proteasoma que participa en el recambio intracelular de las proteínas.
La ubiquitina consta de 76 aminoácidos (8,6 kDa), está formada por cinco láminas y una hélice que es la responsable de la ubiquitinación, proceso estrictamente regulado, según el cual la ubiquitina actúa a modo de etiqueta para que la proteína pueda ser reconocida por el proteasoma para su degradación. Para que se dé este fenómeno, deben producirse dos etapas sucesivas: una de marcación de la proteína diana con numerosas moléculas de ubiquitina, y otra de degradación de la proteína ubiquitinada por el proteasoma 26S, obteniéndose pequeños péptidos de 8 a 9 residuos. Esta propiedad de la ubiquitina, de marcar selectivamente a las proteínas condenadas a desaparecer de escena, es lo que le valió ser denominada como "el beso de la muerte" por la Academia Real Sueca. Si bien la proteína degradada ya no es reutilizada, los aminoácidos que la componían sí.
Por otra parte, los priones (acrónimo de proteína infecciosa) son partículas de naturaleza proteica. Esta definición se ha empleado para designar a los patógenos que inducen algunas alteraciones neurológicas en los vertebrados, como la encefalopatía espongiforme bovina, el prurito lumbar o scrapie de las ovejas y cabras o la enfermedad de Creutzfeldt-Jakob en el hombre. Todas estas encefalopatías se caracterizan por una vacuolización de la materia gris del sistema nervioso central (carácter espongiforme), por la acumulación de fibras proteicas en las células nerviosas y por ser transmisibles. El término prión fue propuesto por Stanley Prusiner (premio Nobel de Medicina en 1997), según el cual, los priones son estructuras capaces de autorreplicarse a formas infecciosas, mediante cambios conformacionales, y serían la causa de las encefalopatías espongiformes transmisibles o prionopatías.
Defectos en el metabolismo proteico y en la vía de la ubiquitina son causas bien conocidas de otras enfermedades neurodegenerativas, como la enfermedad de Parkinson, pero no se les había implicado previamente en la encefalopatía espongiforme. Ahora, algunos resultados ya sugieren la posibilidad de que anomalías en el mecanismo de la ubiquitina tengan funciones importantes para todas las encefalopatías espongiformes. De hecho, los científicos aseguran que lo anterior podría mejorar la comprensión de cómo los priones destruyen los cerebros de seres humanos, del ganado vacuno y de ovejas infectadas.
Para la medicina actual, todos estos avances tienen una gran trascendencia, especialmente en la prevención dirigida a determinados elementos ambientales específicos que pueden ser factores de riesgo en la predisposición detectada. La profilaxis puede hacerse sobre modos y estilos de vida complementados en ocasiones con medidas farmacológicas que, por ejemplo, bloqueen receptores de factores patógenos identificados en cada individuo. Y si la enfermedad aparece, combatirla con medicamentos específicos para una determinada diana, lo que constituye la moderna farmacogenética.
Priones, Proteasomas y vacas locas
Todas las células tienen la capacidad de degradar selectivamente las proteínas intracelulares mal desplegadas estructuralmente, que, si se acumulan, podrían interferir con la función normal y ser tóxicas para el organismo. Tales proteínas pueden presentarse por mutaciones, errores en la expresión génica, fallas de desdoblamiento, desnaturalización espontánea o por daño después de su síntesis (por ejemplo, debido a la acción de los radicales libres del oxígeno). Cuantas veces tales acontecimientos ocurren en las células es incierto, en gran parte porque la vía ubiquitina-proteasoma degrada rápidamente tales proteínas aberrantes, incluyendo los que causan varias enfermedades hereditarias, como la fibrosis cística y ciertos hemoglobinopatías.
Este mecanismo también protege contra enfermedades neurodegenerativas. Las características de la esclerosis lateral amiotrófica, enfermedad de Parkinson, demencia de Lewy, enfermedad de Huntington y la enfermedad de Alzheimer pueden variar ampliamente, pero todas se caracterizan por la presencia de proteínas anormales en las inclusiones intracelulares que se asocian generalmente a la ubiquitina y a los proteasomas. Se ha observado que estas patologías resultan de la acumulación de especies proteicas que son tóxicas, aunque crece la evidencia que estas inclusiones amiloideas son un correlativo más bien que la causa inmediata de la enfermedad y que la alteración neuronal resulta de la presencia de pequeños micro agregados solubles de la proteína anómala.
El mecanismo por el cual estas proteínas similares a las partículas amiloideas interfieren con la función normal y eventualmente promueven la apoptosis es todavía desconocido. Una hipótesis que frecuentemente se ha propuesto es que la vía ubiquitina-proteasoma normalmente elimina estos elementos tóxicos mal desplegados, pero que igualmente y de forma gradual se acumulan, y eventualmente, especialmente en personas mayores, colapsan la capacidad de este sistema proteolítico e interfieren con su funcionamiento. La acumulación posterior de estas proteínas finalmente causa una falla de los mecanismos proteolíticos de las neuronas que son necesarios para la función normal y la supervivencia de la célula. Respecto a esto, ha crecido de forma importante la información directa que las proteínas anormales en estas enfermedades pueden inhibir la funcionalidad del proteasoma.
Recientemente, sin embargo, Kristiansen y colaboradores, presentaron evidencia contundente respecto a que los agregados solubles de proteínas tóxicas pueden causar la enfermedad por prión, específicamente por la inhibición de la función del proteasoma 26S. Las enfermedades inducidas por priones son un sistema de enfermedades neurodegenerativas transmisibles y fatales que están asociadas a la encefalopatía espongiforme, incluyendo enfermedades que ocurren en los seres humanos, la enfermedad de Creutzfeldt-Jakob, el síndrome de Gerstmann-Sträussler-Scheinker y el kuru; en ovejas, scrapie; y en vacas, la encefalopatía espongiforme bovina (o “enfermedad de las vacas locas”). Más de 150 personas en Gran Bretaña han muerto de una variante de la enfermedad de Creutzfeldt-Jakob adquirida con la ingestión de carne de vacuno que contenía el prión. Estas patologías habían representado un importante misterio biológico, sobre todo porque se había demostrado que el agente transmisor carecía de ácidos nucleicos, pero los estudios seminales de Stanley Prusiner y de Charles Weissmann establecieron que las alteraciones eran causadas por una forma aberrante de la proteína del prión. Esta proteína es capaz de incorporarse a las células e inducir un cambio conformacional de la forma soluble normal de la proteína del prion (PrPc) en una especie transmisiblemente tóxica (PrPsc). En esta conformación patógena, el prión es sobre todo insoluble, es resistente a las proteasas o los detergentes, tienen la estructura beta amiloidea, y posee agregados más grandes que la especie normal. Este cambio de conformación y la acumulación resultante de PrPsc causan en última instancia la severa pérdida neuronal, gliosis y el aspecto espongiforme. Por consiguiente, los ratones que no expresan la proteína normal no son susceptibles a la enfermedad.
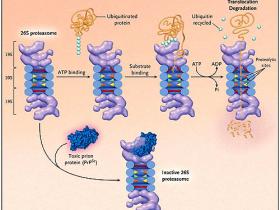
Una importante brecha en nuestra comprensión ha sido dilucidar cómo la conversión de PrPc a PrPsc elimina a las neuronas. Para clarificar el mecanismo neurotóxico, Kristiansen y colaboradores investigaron si PrPsc interfiere con la función del proteasoma, usando acercamientos in vivo e in vitro. En el sistema ubiquitina-proteasoma, las proteínas son rápidamente degradadas mediante el acoplamiento covalente a una de las cadenas de la ubiquitina, que las marca para que sean hidrolizadas por el complejo proteasoma 26S. Cuando ya están debidamente marcadas con una de las cadenas de la ubiquitina, los substratos las atan al componente regulador de los proteasomas 19S, que desmonta las cadenas de ubiquitina y recicla las moléculas de ubiquitina. Entonces, con un proceso dependiente de ATP que recién ahora se comienza a comprender, las ATPasas de la partícula 19S despliegan el substrato y lo desplazan a través de un estrecho canal ubicado en el proteasoma 20S para la degradación (figura 1). Dentro de esta partícula cilíndrica hueca, la proteína “condenada” se une a pequeños péptidos, y entonces se libera del proteasoma y es rápidamente degradada en aminoácidos por las peptidasas citosólicas.
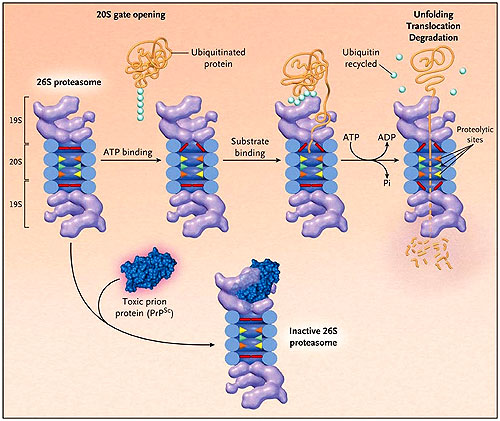
La degradación de la proteína por el proteasoma 26S implica múltiples pasos que dependen de la presencia de ATP, incluyendo la abertura del canal bloqueado para la entrada del substrato en la partícula 20S, la unión del substrato ubiquitinado, el desmontaje de la cadena de ubiquitina y el desplazamiento de la proteína anómala a través del anillo de la ATPasa y del canal. La unión de los oligómeros de la proteína tóxica del prion (PrPsc) a las ATPasas puede explicar las recientes observaciones de Kristiansen y colaboradores que en las células infectadas por PrPsc, se inhibe la degradación de la proteína y de los substratos del péptido por los proteasomas y que las proteínas ubiquitinadas (círculos azules) se acumulan. El ATP señala a la adenosina trifosfato, el ADP a la adenosina difosfato y Pi al fosfato inorgánico.
Kristiansen y colegas demostraron que las neuronas y las células de neuroblastoma infectadas con priones presentan una reducida actividad proteasomal contra los substratos. El proteasoma 20S contiene dentro de su cámara central seis sitios proteolíticos. Utilizando substratos específicos para cada sitio, el grupo de Kristiansen demostró que tres actividades se encontraban minimizadas en las neuronas infectadas con priones. Además, observaron que en estas neuronas infectadas, PrPsc estaba presente en el citosol y que las células tenían una menor capacidad para degradar las proteínas fluorescentes transfectadas, las cuales son substratos de la vía ubiquitina-proteasoma. Por otra parte, al “curar” estas células de los priones se restauraban los índices de degradación.
Al inocular los ratones transgénicos con priones, los mismos autores demostraron que PrPsc induce similares defectos en la capacidad del cerebro para degradar la proteína fluorescente. En las neuronas infectadas de estos ratones, los depósitos ubiquitinados acumulados en el citosol se asemejaban mucho a los encontrados en enfermedades neurodegenerativas de los humanos y en las células tratadas con inhibidores farmacológicos del proteasoma. Aunque la menor capacidad de degradación es consistente con la pérdida de actividad proteasomal, también podría deberse a defectos en la ubiquitinación o en otros pasos de la vía metabólica. Sin embargo, los autores obtuvieron más datos de apoyo al utilizar los proteasomas purificados 26S y PrPsc aislados de cerebros infectados de ratón y de ratones recombinantes para PrP semejante al PrPc nativo. Una clara reducción en la actividad de los proteasomes purificados (así como también en las neuronas) ocurrió solamente con las formas agregadas de PrPsc. Esta inhibición fue resultado de una asociación estequiométrica de las especies oligoméricas con los proteasomas.
Aunque ahora ya se sabe que el PrPsc citosólico puede interferir con las actividades de la peptidasa de los proteasomas y que las neuronas infectadas son defectuosas para la degradación de la proteína, no existen pruebas que éste sea el único mecanismo tóxico, o que específicamente conduzca a la pérdida neuronal en las enfermedades priónicas. La pérdida de la función proteasomal afecta a muchos procesos celulares críticos y puede inducir apoptosis. Además de ser un sistema de control de calidad, esta vía es primordial en la regulación de las redes de señalización celular y en los mecanismos metabólicos, alcanzando una “exquisita” selectividad debido a la presencia en los seres humanos de centenares y diversas ligasas de ubiquitina que participan en la eliminación de muchas proteínas. Por lo tanto, no es sorprendente, que en los modelos celulares para todas las enfermedades neurodegenerativas, el tratamiento con inhibidores de proteasoma promueva la patogénesis y la muerte neuronal. Los sitios proteolíticos de los proteasomas 20S son el blanco del medicamento anticancerígeno denominado bortezomib, el que es ampliamente utilizado para el tratamiento del mieloma múltiple, un cáncer que parece ser particularmente dependiente de los proteasomas para la supervivencia y para la degradación de las inmunoglobulinas. Una característica importante de su selectividad es que no se incorpora al sistema nervioso central, donde probablemente podría acelerar el inicio de la enfermedad neurodegenerativa.
Tampoco se ha podido responder exactamente cómo los pequeños agregados de PrPsc inhiben la hendidura de los péptidos. La “decisión” de qué proteínas se desplazan en la partícula y cuáles se degradan depende del anillo de ATPasas en el componente regulador 19S, el cual es probablemente el sitio en donde los oligomeros de PrPsc se unen (figura 1). Normalmente, las proteínas celulares que son destinadas a degradación son desdobladas por estas ATPasas antes de ser desplazadas dentro de la partícula 20S. Sin embargo, la excepcional estabilidad de PrPsc hace inverosímil que estas estructuras se puedan desmontar de igual forma, y probablemente los micro agregados de PrPsc son demasiado grandes para atravesar el anillo de ATPasas más estrechos y los poros bloqueados en la partícula 20S. Así, pueden comportarse como un “corcho pegajoso” que interfiere con la entrada de otros substratos en la partícula 20. Un mecanismo tan simple que ahora afortunadamente se puede evaluar.
Fuente bibliográfica
On Prions, Proteasomes, and Mad Cows
Alfred L. Goldberg, Ph.D.
Department of Cell Biology, Harvard Medical School, Boston, USA.
N Engl J Med. 2007 Sep 13; 357(11):1150-2