















 41 / 62
41 / 62

C o n t e n i d o d i s p o n i b l e e n h t t p : / / www. n e umo l o g i a - p e d i a t r i c a . cl
39
Neumol Pediatr 2016; 11 (1): 38 - 43
Tratamiento de fibrosis quística: pasado y presente
APOYO NUTRICIONAL Y ADMINISTRACIÓN DE ENZIMAS
DIGESTIVAS
(4)
El buen estado nutricional influye positivamente tanto
en la calidad de vida como en la sobrevida. Se recomienda
administrar una dieta que proporcione 120-140% de las
calorías diarias recomendadas, alta en proteínas y que no
esté restringida en grasas u otros ingredientes, ricas en sal,
vitaminas, minerales y con aporte de enzimas pancreáticas si
es necesario. Cuando la ganancia de peso y crecimiento son
insuficientes, se recomienda aumentar el aporte nutricional (±
20% en promedio). Si la relación peso/talla persiste inferior al 90
% del ideal o si hay clara disminución en la curva de crecimiento
se recomienda colocar una sonda nasogástrica o gastrostomía
para alimentación enteral nocturna continua.
La ingestión de altas concentraciones de enzimas
pancreáticas es un factor de riesgo para el desarrollo de
colonopatía fibrosante y no se recomienda exceder dosis de
10.000 unidades de lipasa / kg / día. Los pacientes que no estén
bien controlados con dosis suficientes de enzimas deben ser
evaluados por otras causas de malabsorción y además evaluar
el beneficio de agregar inhibidores de ácido gástrico.
La terapia de reemplazo de enzimas pancreáticas debe
ser individualizada y adaptada a cada comida. Dependiendo de la
duración de cada comida las enzimas se administran al comienzo
o se dividen en una mitad o 2/3 al principio y un tercio o mitad
al final. Se debe evitar masticar las cápsulas, ya que liberaría
enzimas en la cavidad bucal que pueden dañar la mucosa, si
es necesario abrir las cápsulas se pueden administrar con jugo,
agua o puré de manzana, evitando líquidos alcalinos.
Aún sigue siendo controvertida la suplementación con
taurina y ácido ursodesoxicólico que mejoran la función hepática
pero no está claro si esto tiene un impacto positivo en el curso
de la enfermedad.
Los pacientes con FQ tienen riesgo de perder
cantidades significativas de sal a través del sudor, especialmente
en climas más cálidos y los lactantes, los que deberían recibir
suplementos de sal. Debido a la insuficiencia pancreática y la
mala absorción de grasa se reduce la absorción de las vitaminas
liposolubles (A, D, E y K) y la suplementación se considera un
estándar de atención en todos los pacientes con insuficiencia
pancreática. El papel de otros suplementos como antioxidantes y
ácidos grasos omega- 3 sigue siendo poco claro.
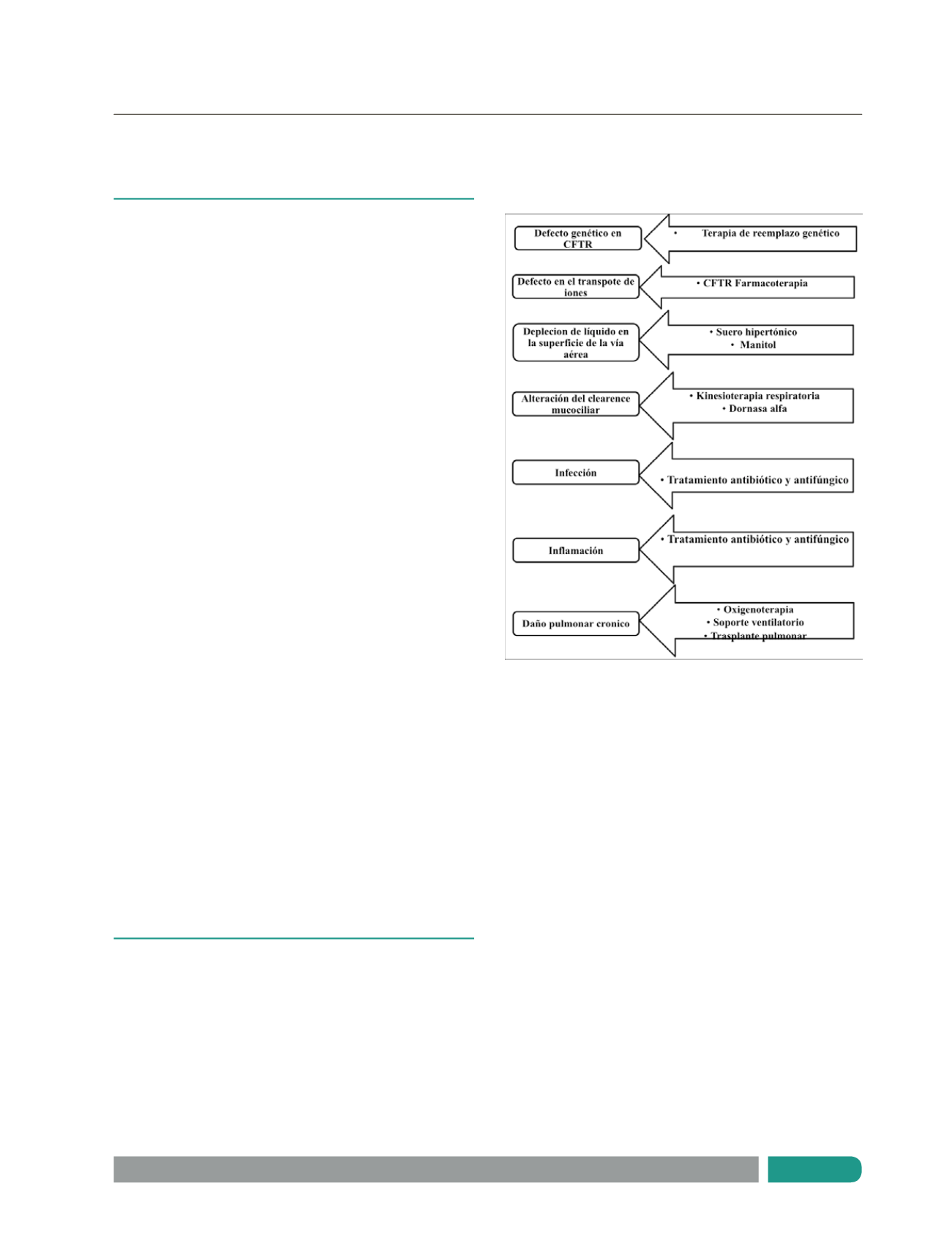
TRATAMIENTO DE LA ENFERMEDAD PULMONAR
En la Figura 1 se muestran las distintas terapias y su
correlación con la fisiopatología de la enfermedad pulmonar.
Terapia de reemplazo génica y farmacoterapia
La terapia génica implica la inserción de una copia de ADN
que codifica un regulador de transmembrana de la fibrosis quística
(CFTR) normal dentro de las células respiratorias defectuosas
utilizando diferentes vectores (5). Ningún estudio ha podido demostrar
un efecto a largo plazo, si bien estos estudios siguen en curso; el
enfoque actual está dirigido a la farmacoterapia (6).
El objetivo de la farmacoterapia es mejorar el tráfico,
expresión o función de CFTR (7). Para pacientes portadores de
mutaciones clase I el tratamiento con ataluren, un compuesto
que promueve la lectura a través de codones truncados
prematuramente en el ARNm de CFTR, ha demostrado aumentar
la expresión de CFTR. Sin embargo, los ensayos clínicos fase 3
han demostrado beneficios sólo en un subgrupo de pacientes.
En la Figura 2 se grafica los niveles de cloro en sudor
en relación a la actividad de CFTR.
Para pacientes con la mutación G551D (clase III)
ivacaftor fue aprobado por la FDA el 2012 para su uso en
mayores de 6 años. Ivacaftor es un potenciador de la función, que
activa el CFTR defectuoso en la superficie celular. En pacientes
portadores de al menos una copia de la mutación G551D,
ivacaftor administrado por vía oral, mejoró la función pulmonar,
redujo las exacerbaciones pulmonares, se asoció a aumento de
peso y mejoría de los síntomas respiratorios. También disminuyó
significativamente las concentraciones de cloruro en el test del
sudor lo que refleja el impacto de la droga en el defecto básico
en la FQ. La experiencia a largo plazo sigue siendo limitada.
Recientemente, estudios han demostrado una eficacia similar
en pacientes con otras mutaciones clase III, siendo su uso
fuertemente recomendado en los pacientes portadores al menos
1 copia de la mutación G551D (12).
En pacientes con la mutación
Δ
F508, lumicaftor, un
“corrector” de CFTR, ha sido diseñado para mover la proteína de
Figura 1.
Fisiopatología de la fibrosis quística y
objetivos del tratamiento