Un modelo para una común insuficiencia cardíaca
La insuficiencia cardíaca con fracción de eyección preservada (ICFEp) es un síndrome frecuente con alta morbilidad y mortalidad para el que no existen tratamientos basados en pruebas. Sin embargo, en un reciente estudio se ha dado a conocer que el estrés metabólico e hipertensivo concomitante en ratones, provocado por la combinación de una dieta rica en grasas y la inhibición de la óxido nítrico sintasa constitutiva, recapitula las numerosas características sistémicas y cardiovasculares de la ICFEp ocurrida en humanos. La expresión de uno de los efectores de respuesta a proteínas desplegadas, la variante de splicing de la proteína de unión a X-box 1 (XBP1s), se redujo en el miocardio del modelo de roedores y en humanos con ICFEp. Mecanísticamente, esta disminución resultó en el aumento de la actividad de la óxido nítrico sintasa inducible (iNOS) y la S-nitrosilación de la endonucleasa que requiere inositol 1α (IRE1α), culminando en un empalme XBP1 defectuoso. Además, la supresión farmacológica o genética de iNOS, o la sobreexpresión de XBP1s restringida a cardiomiocitos, mejoraron el fenotipo de la ICFEp. Por lo tanto, la desregulación impulsada por iNOS de la vía IRE1α-XBP1 es un mecanismo crucial en la disfunción cardiomiocitaria en este tipo de insuficiencia cardíaca.
Inhibición de la respuesta a proteínas desplegadas
La insuficiencia cardíaca es una enfermedad letal que afecta al menos a 26 millones de personas en todo el mundo. Sin embargo, falta un modelo preclínico que recapitule fielmente la forma más común de este síndrome, conocida como insuficiencia cardíaca con fracción de eyección preservada (ICFEp). En un reciente estudio Gabriele Schiattarella y colaboradores (DOI: 10.1038/s41586-019-1100-z) reportan el desarrollo de un modelo de ratón para el estuio de la ICFEp, y su utilidad para identificar una vía molecular que no se conocía anteriormente y que desempeña un papel clave en esta enfermedad.
La insuficiencia cardíaca se ha visto generalmente como una incapacidad del corazón para bombear la suficiente sangre para satisfacer las necesidades metabólicas. El debilitamiento del músculo cardíaco se cuantifica midiendo la reducción en el porcentaje de sangre en el corazón expulsada con cada latido, lo que se denomina fracción de eyección. Este síndrome se conoce como insuficiencia cardíaca con fracción de eyección reducida (ICFEp). Pero en más de la mitad de los casos de insuficiencia cardíaca, la fracción de eyección sigue siendo normal, aunque las anomalías en la contracción del músculo cardíaco se pueden detectar con pruebas más sensibles. Esta forma de insuficiencia se define por el deterioro del llenado del corazón con sangre. La disfunción en otros órganos, incluyendo pulmones, riñones y músculo esquelético, también es probable que contribuya a los síntomas de la enfermedad.
En la ICFEp, las células endoteliales que recubren los pequeños vasos sanguíneos del corazón se vuelven disfuncionales y no logran sintetizar cantidades adecuadas de óxido nítrico (NO), una sustancia implicada en la relajación de los vasos sanguíneos. Además, las células del tejido conectivo llamadas fibroblastos generan cicatrices. Y los propios cardiomiocitos se vuelven más rígidos debido a los cambios relacionados con las proteínas implicadas en las vías de señalización molecular y la contracción celular. El resultado final es un llenado lento del corazón.
Ninguna de las terapias existentes es de utilidad para la ICFEp, y actualmente no existe un buen modelo animal para estudiarla. La modelización de la ICFEp es un reto debido a la compleja interacción de muchos factores contribuyentes y a la plétora de manifestaciones clínicas de la enfermedad. En lugar de recrear todos los estímulos que conducen a la ICFEp, Schiattarella y sus colegas combinaron dos factores de riesgo principales: la obesidad relacionada con la intolerancia a la glucosa (una incapacidad para transportar el azúcar de forma eficiente desde la sangre a las células, lo que puede conducir a la diabetes) y la hipertensión arterial, en ratones. El razonamiento de los autores fue que cada uno de estos factores activa varias vías asociadas con la enfermedad.
Los ratones se volvieron obesos e intolerantes a la glucosa con una dieta alta en grasas. La hipertensión arterial fue inducida por la administración de un fármaco llamado Nω-nitro-L-arginina metil ester (L -NAME), que inhibe las enzimas que normalmente se expresan a niveles constantes para catalizar la producción de NO (llamadas NO sintasas constitutivas). Los ratones tratados de esta manera tenían varias características de ICFEp, incluyendo el llenado deficiente del corazón, la incapacidad para hacer ejercicio, congestión pulmonar y aumento de los niveles de marcadores moleculares de inflamación en corazón y sangre (figura 1). En particular, la fracción de eyección en estos ratones permaneció normal hasta por lo menos los 12 meses de edad, que es la mitad de la vida de estos animales.
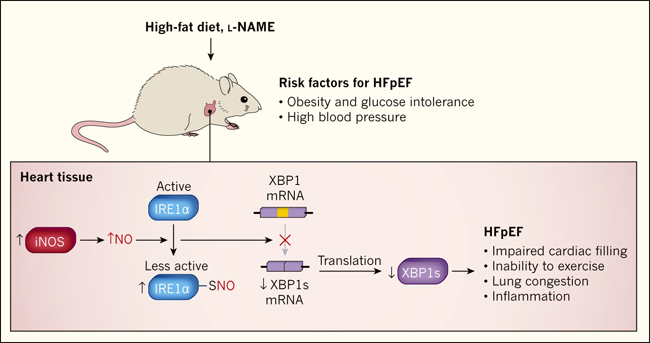
Figura 1. La inhibición de la respuesta a proteínas desplegadas contribuye a la insuficiencia cardíaca con una fracción de eyección preservada.
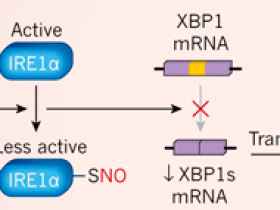
Schiattarella y colaboradores (DOI: 10.1038/s41586-019-1100-z) alimentaron a ratones con una dieta alta en grasas para producir obesidad e intolerancia a la glucosa (una incapacidad para transportar el azúcar eficientemente de la sangre a las células, lo que puede llevar a la diabetes), y les suministró el fármaco Nω-nitro-L-arginininina metil éster (L-NAME), el cual causó presión arterial elevada. Estos tratamientos indujeron algunas de las características de la insuficiencia cardíaca con fracción de eyección preservada (HFpEF, por sus siglas en inglés), incluyendo el deterioro del llenado del corazón, incapacidad para hacer ejercicio, congestión pulmonar y aumento de los niveles de marcadores moleculares asociados con la inflamación. Los autores también observaron un aumento de la expresión de la enzima óxido nítrico sintasa inducible (iNOS), que aumenta la producción de óxido nítrico (NO). La adición de NO a los átomos de azufre (S-nitrosilación) de la enzima que requiere inositol 1α (IRE1α) disminuye la actividad de esta proteína. IRE1α es un componente de la respuesta a proteínas desplegadas (UPR, del inglés unfolded protein response), un mecanismo que protege a las células contra niveles anormales de proteínas mal plegadas. La disminución de la actividad de IRE1α conduce a una reducción de la conversión (empalme) del ARN mensajero codificante de la proteína de unión a X-box 1 (XBP1) al ARN m que codifica la proteína XBP1s generada por empalme alternativo. XBP1 es un factor de transcripción que activa los genes UPR. Los autores observaron que la actividad de empalme de IREα y los niveles de XBP1s se redujeron en su modelo de ratón de ICFEp.
Los autores luego utilizaron su modelo para investigar el papel de las proteínas mal plegadas en la ICFEp. Estas proteínas se acumulan en varios trastornos en los que el estrés cardíaco está presente, incluyendo la ICFEp, y activan un programa molecular conocido como respuesta a proteínas desplegadas (UPR). Schiattarella y sus colegas se centraron en la enzima que requiere inositol 1α (IRE1α), que se activa en respuesta al estrés celular causado por un exceso de proteínas mal plegadas. IRE1α corta el ARN mensajero que codifica la proteína de unión a X-box 1 (XBP1) y la transforma en un ARNm más corto que codifica una proteína llamada XBP1 de empalme o splicing (XBP1s). XBP1s es un factor de transcripción que activa los genes involucrados en la UPR.
Schiattarella y sus colegas encontraron que la activación de IRE1α y los niveles de XBP1s se redujeron en los corazones de los ratones modelo de ICFEp, así como en las muestras de tejido cardíaco de las personas con la condición, en comparación con las de individuos sanos (figura 1). Por el contrario, los niveles de XBP1s cardíacos se incrementaron o no se modificaron en ratones y humanos con ICFEp. La disminución en los niveles de XBP1s en los corazones de los ratones con ICFEp se debió a la modificación de IRE1α por S-nitrosilación. Este proceso, en el que una molécula de NO se adhiere a átomos específicos de azufre en las proteínas, se sabe que disminuye la actividad de la célula.
Aunque los investigadores no identificaron las células que producen NO, observaron que la NO sintasa inducible (iNOS), una enzima que fue altamente expresada en su modelo de ICFEp, promovió la S-nitrosilación. A continuación, demostraron que la inhibición farmacológica o genética de iNOS o la sobreexpresión específica de XBP1s en los cardiomiocitos atenuaba los defectos del llenado cardíaco, la incapacidad para hacer ejercicio y la congestión pulmonar en ratones a los que se les indujo características de la ICFEp. Estos hallazgos destacan el papel de la pérdida de XBP1s en los mecanismos que subyacen a esta enfermedad.
Este trabajo demuestra que dos factores de riesgo comunes para la ICFEp son suficientes para reproducir muchas de las manifestaciones de esta enfermedad. Este modelo de ratón será útil para diseccionar los mecanismos de la enfermedad y desarrollar nuevos tratamientos. Además de ser un animal valioso para el estudio de procesos fisiológicos complejos y probar terapias, el ratón tiene la ventaja de ser fácil de manipular genéticamente y tener un ciclo de vida corto. Este último factor también facilitará el estudio de la influencia del envejecimiento, otro factor de riesgo clave para la ICFEp.
Algunas preguntas siguen sin respuesta. La reversión incompleta de las manifestaciones de la enfermedad observadas en respuesta a la inhibición de la iNOS sugiere que otras vías moleculares también están implicadas en la reducción de los niveles de XBP1s. De manera más general, el estudio explora un único mecanismo candidato para esta enfermedad. Es probable que otros procesos también contribuyan, y que el nuevo modelo de ratón para el estudio de la insuficiencia cardíaca con fracción de eyección preservada proporcione una plataforma in vivo con la cual definirlos.
Fuente bibliográfica
A mouse model for the most common form of heart failure
Dulguun Amgalan & Richard N. Kitsis
Departments of Medicine (Cardiology) and Cell Biology, and in the Wilf Family Cardiovascular Research Institute, Albert Einstein College of Medicine, Bronx, New York.
DOI: 10.1038/d41586-019-00983-4