Ateroesclerosis: cĂ©lulas que desafĂan la muerte
La aterosclerosis es el proceso patolĂłgico que subyace a un ataque cardĂaco y accidente cerebrovascular. Se caracteriza por la acumulaciĂłn de cĂ©lulas vasculares anĂłmalas y restos celulares apoptĂłticos que no pueden ser removidos por razones aĂşn desconocidas. En una reciente investigaciĂłn, se observĂł que la aterogĂ©nesis se vincula con una regulaciĂłn positiva de CD47, una molĂ©cula anti-fagocĂtica clave que confiere señales de resistencia a cĂ©lulas anormales frente a la eliminaciĂłn fagocĂtica de este material celular. Notablemente, a travĂ©s del uso de anticuerpos que bloquean a la proteĂna CD47, se logrĂł revertir este defecto, disminuyendo la inflamaciĂłn, normalizando el tejido vascular y aminorando la aterosclerosis en varios modelos de estudio en ratones. Este enfoque actualmente está siendo evaluado en ensayos clĂnicos para pacientes con trastornos inflamatorios crĂłnicos lo que podrĂa contribuir a la reducciĂłn de las crecientes tasas de enfermedades cardiovasculares.
Restos celulares apoptĂłticos
Los ataques cardĂacos y los accidentes cerebrovasculares, que son las principales causas de muerte en todo el mundo, comienzan con un proceso llamado aterosclerosis, en el que se forman placas - acumulaciones de lĂpidos, cĂ©lulas, matriz extracelular y restos celulares- en determinadas zonas de las arterias. A pesar de que las arterias de la mayorĂa de las personas contienen muchas de estas placas, sĂłlo un pequeño porcentaje causa enfermedad. En un reciente artĂculo Yoko Kojima y colegas (Nature. 2016 Aug 4;536(7614):86-90) proporcionan un mecanismo plausible que podrĂa explicar por quĂ© algunos casos se convierten en un peligro clĂnico.
Una caracterĂstica clave de las placas que revisten peligro clĂnico es una estructura llamada centro necrĂłtico, el que contiene cĂ©lulas muertas que han sufrido un tipo de muerte celular conocido como necrosis. Este core necrĂłtico se caracteriza por estar inflamado y por tener una capa fibrosa delgada que cubre la placa y la separa del lumen central de la arteria (figura 1). Cuando se rompe el casquillo o se erosiona, el material necrĂłtico se expone a las plaquetas, necesarias para la coagulaciĂłn de la sangre. Esta exposiciĂłn da lugar a la agregaciĂłn plaquetaria (conocida como trombo), que puede bloquear el vaso sanguĂneo y por lo tanto provocar un ataque al corazĂłn o un derrame cerebral al privar al corazĂłn o al cerebro de oxĂgeno. El nĂşcleo necrĂłtico, que alberga restos celulares inflamatorios, promueve la disrupciĂłn de la capa al contribuir a la degradaciĂłn de sus proteĂnas estructurales, como el colágeno, además de generar estrĂ©s fĂsico en esta estructura. Por lo tanto, la comprensiĂłn sobre cĂłmo se desarrolla el nĂşcleo necrĂłtico es un objetivo urgente en la investigaciĂłn de patologĂas cardĂacas.
Para determinar cĂłmo las cĂ©lulas moribundas sufren necrosis en las placas de ateroma, es necesario entender cĂłmo el cuerpo evita este tipo de muerte celular. Miles de millones de cĂ©lulas en el organismo mueren cada dĂa a travĂ©s de un proceso denominado apoptosis, que inicialmente evita la ruptura de la membrana celular y la fuga del contenido inflamatorio celular. Las cĂ©lulas apoptĂłticas se eliminan rápidamente y de forma segura mediante un proceso conservado evolutivamente que involucra la remosiĂłn de restos celulares mediante cĂ©lulas fagocĂticas (efferocytosis), en el que los cuerpos apoptĂłticos son interiorizados y destruidos antes de que se produzca la lisis de sus membranas.
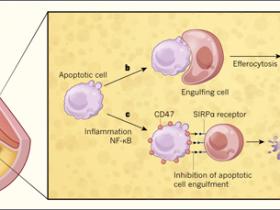
La fagocitosis de cuerpos apoptĂłticos requiere la señalizaciĂłn entre la cĂ©lula que muere y el fagocito: factores producidos por la cĂ©lula apoptĂłtica promueven la migraciĂłn de fagocitos hacia cĂ©lulas apoptĂłticas, y el despliegue de marcadores  permiten el reconocimiento en la superficie de cĂ©lulas apoptĂłticas a travĂ©s del acoplamiento a receptores presentes en los fagocitos. Como mecanismo de seguridad, las cĂ©lulas sanas expresan a menudo molĂ©culas dispuestas en su superficie que impiden este proceso de reconocimiento para impedir ser internalizados por fagocitos. La proteĂna CD47 es un ejemplo de una molĂ©cula de este tipo, la que actĂşa a travĂ©s del receptor SIRPα en los fagocitos, de modo de inhibir su eliminaciĂłn.
Pero, Âżque ocurre en las placas? Los estudios han demostrado que la fagocitosis de restos celulares se encuentra deteriorada en las placas de pacientes que no han alcanzado la etapa vulnerable, y los experimentos que han utilizado ratones genĂ©ticamente modificados han demostrado una relaciĂłn causal entre el fallo de este mecanismo y la necrosis de la placa. Por lo tanto, en las placas consideradas como avanzadas, el debris celular sin removerse eventualmente se convierten en fugas de molĂ©culas potencialmente dañinas, lo que resulta en un proceso llamado necrosis secundaria.Â
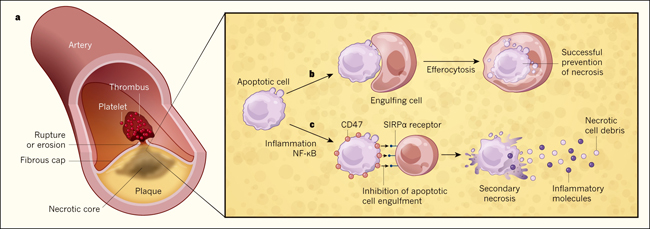
Figura 1. La defectuosa eliminaciĂłn de cĂ©lulas muertas puede contribuir a la formaciĂłn de placas aterosclerĂłticas clĂnicamente peligrosas.
a, muchas placas que revisten un riesgo clĂnico contienen una estructura llamada nĂşcleo o core necrĂłtico, caracterizado por la inflamaciĂłn y la muerte celular necrĂłtica. En la aterosclerosis, si la capa fibrosa que cubre la placa se rompe o se erosiona, la liberaciĂłn de material del nĂşcleo necrĂłtico puede desencadenar la agregaciĂłn de plaquetas (conocido como trombo) y el bloqueo arterial, lo que finalmente puede resultar en un ataque al corazĂłn o un derrame cerebral. La comprensiĂłn de cĂłmo las placas desarrollan un estado necrĂłtico es una materia de vital importancia. b, las cĂ©lulas de las placas se someten a un tipo de muerte celular no inflamatoria denominada apoptosis. En estas placas asintomáticas no necrĂłticas, la rápida eliminaciĂłn de restos apoptĂłticos mediada por fagocitos evita la necrosis. c, Yoko Kojima y colegas encontraron que las condiciones inflamatorias de una aterosclerosis avanzada conducen a la expresiĂłn persistente de CD47 en las cĂ©lulas de la placa, a travĂ©s del mediador proinflamatorio NF-κB. Cuando estas cĂ©lulas se vuelven apoptĂłticas, CD47 envĂa una señal a travĂ©s del receptor SIRPα en la superficie celular de fagocitos para bloquear la internalizaciĂłn. Entonces, las cĂ©lulas no fagocitadas se someten a un tipo de muerte celular llamada necrosis secundaria, lo que lleva a la liberaciĂłn de molĂ©culas inflamatorias y a la formaciĂłn de nĂşcleos necrĂłticos de restos celulares.
La naturaleza compleja de la aterosclerosis y de la fagocitosis de cuerpos apoptĂłticos sugiere que mĂşltiples mecanismos causan defectos a medida que progresa la formaciĂłn de placas. Los investigadores mostraron previamente que las cĂ©lulas muertas en la placa exhiben un dĂ©ficit en la expresiĂłn de la señal "no me comas" correspondiente a la proteĂna calreticulina. Por otra parte, la proteĂna receptora MerTK presente en los macrĂłfagos fagocĂticos que ejercen su funciĂłn en las placas avanzadas, sufre una degradaciĂłn en las mismas condiciones inflamatorias antes descritas. La proteasa ADAM17 activa al factor de necrosis tumoral alfa (TNF-α), el que induce a CD47 en cĂ©lulas vasculares del mĂşsculo liso, y tambiĂ©n promueve la esciciĂłn de MerTK10. Tanto la activaciĂłn de ADAM17 y la ruptura de MerTK se han involucrado en la progresiĂłn de placas hacia estados clĂnicamente peligrosos.Â
El tratamiento con anticuerpos anti-TNF-alfa podrĂan potencialmente bloquear la inducciĂłn de CD47, y esta estrategia ha sido exitosa en debilitar enfermedades autoinmunes en las que TNF-α es un gatillador principal, como por ejemplo en la artritis reumatoide. Sin embargo, en la aterosclerosis,es probable que la inflamaciĂłn se produzca a travĂ©s de mĂşltiples vĂas. Otra preocupaciĂłn es que el tratamiento anti-TNF-α puede comprometer las defensas, lo que pondrĂa en entredicho su uso a largo plazo como medida preventiva en la mayorĂa de las personas asintomáticas que se encuentran en riesgo de sufrir un episodio cardĂaco agudo.
El uso de anticuerpos anti-CD47, que está siendo probado como un tratamiento para el cáncer en ensayos clĂnicos tempranos, presenta otros retos. CD47 es utilizado por los glĂłbulos rojos para prevenir su internalizaciĂłn prematura antes de la senescencia celular, y un efecto adverso importante de la terapia anti-CD47 es la anemia. Además, CD47 tiene funciones en la adhesiĂłn celular y la migraciĂłn, por lo que su inhibiciĂłn puede causar efectos adversos relacionados con la formaciĂłn de vasos sanguĂneos y las defensas inmunolĂłgicas.
Otra estrategia terapéutica se basa en la observación de que muchos de los procesos que generan placas vulnerables, pueden ser causados por defectos en un programa biológico conocido como resolución de la inflamación, que normalmente termina una respuesta inflamatoria cuando ya no es necesaria, e inicia la reparación tisular.
La administraciĂłn de compuestos que median este programa de resoluciĂłn ha demostrado ser beneficioso en muchos modelos preclĂnicos de patologĂas con resoluciones defectuosas. Por ejemplo, tal terapia puede mejorar la remociĂłn de restos celulares y suprimir la necrosis en la placa de aterosclerosis avanzada. Por otra parte, la terapia mediadora de la resoluciĂłn inflamatoria puede potenciar la defensa del hospedero y este enfoque ya se está probando en los primeros ensayos clĂnicos orientados hacia condiciones inflamatorias crĂłnicas. Estos y futuros desarrollos basados en el trabajo de Kojima y sus colegas podrán algĂşn dĂa proporcionar una manera segura para mantener sin riesgo a las personas afectadas.
Fuente bibliográfica
Heart disease: Death-defying plaque cells
Ira Tabas
Departments of Medicine, Pathology and Cell Biology, and Physiology, Columbia University School of Medicine, New York, New York 10032, USA.
doi:10.1038/nature18916